
RSS-Feed abonnieren
DOI: 10.1055/a-1384-4641
Rare Diseases of the Orbit
Artikel in mehreren Sprachen: deutsch | English
- 1 General Introductory Remarks
- 2. Interdisciplinarity and “What is rare, What is frequent?” – A Dilemma?
- 3. History-taking and Diagnostics
- 4. Structural Lesions of the Orbit
- 4.4 Bone anomalies and mesodermal defects
- 5. Inflammatory Diseases of the Orbit
- 5.1 Infectious orbital inflammation
- 5.2 Non-infectious orbital inflammation
- 5.2.3 Vasculitis of the orbit
- 6. Degenerative Diseases: Orbital Amyloidosis
- 7. Tumors of the Orbit 2 201 233
- 7.2 Particularities
- 7.2.2 Neoplasms of the orbit (without lacrimal gland neoplasms)
- Literatur
Abstract
This article provides an overview of rare orbital diseases. Congenital malformations, inflammatory diseases, benign and malignant neoplasias are described. Although it represents a relatively small area of the body the orbit contains multiple different tissues. Therefore, a great variety of diseases can be found within the orbital space. That is the reason, why both the completeness and the level of detail in the description of particular diseases must be somewhat limited. Nevertheless, clinical manifestations, important aspects of diagnosis, treatment strategies, and, when specific data are available, the prognosis are described. The authors tried to highlight the most characteristic aspects of the different diseases to describe their relevant aspects in spite of the brevity of the subsections.
#
1 General Introductory Remarks
Oto-rhino-laryngologists and ophthalmologists regularly have to deal with diseases of the orbit. The complex anatomy of this small region of the body and the multitude of different tissues it encompasses entail a high heterogeneity of pathologies. In addition to numerous systemic diseases associated with the orbit, various malformations can arise from a complex embryology, and treatment of orbital diseases must be considered a prime example for interdisciplinary cooperation. The central facial position and close topographic association with the nose and paranasal sinuses, skull base, cranial skull, and temporal region can even require interdisciplinary surgical care.
The orbit serves as protection for the eye and its adnexae. The relatively stable bony frame with the corpus adiposum orbitae provides protection, but because of the confined space, volume expansion can rapidly lead to problems. These features are responsible for the fact that most pathological processes of the orbit primarily manifest ophthalmologically. Exophthalmos, diplopia, and loss of vision are the most frequent clinical signs, and ophthalmologists play a crucial role in detecting and managing these conditions.
Many orbital diseases, such as endocrine orbitopathy, are well known yet still can follow atypical and rare courses. Other conditions are truly rare and so numerous that a complete overview of them all is beyond the scope of this manuscript. Most clinicians likely will never encounter many of the diseases described here yet will still find this comprehensive series of synopses useful. Our intention for this article is to sharpen the clinical perspective and provide preparation for first encounters with possibly unfamiliar manifestations.
In addition, this article does not deal with periorbital (preseptal) diseases. Thus, it does not cover pathologies of the eyelids and the draining lacrimal ducts or traumatology of the orbit. Furthermore, solely ocular diseases are not within the scope of this overview, as these conditions expand beyond the ocular border structures (e. g., to the sclera) or cause concomitant orbital reactions.
#
2. Interdisciplinarity and “What is rare, What is frequent?” – A Dilemma?
Different disciplines vary in perspective on the orbit and perception of the incidence of related conditions, as is also reflected in the literature. Because pathogenic processes related to the orbit are not common, precise incidences are difficult to pinpoint in the different patient populations treated or co-treated within different disciplines. Furthermore, large randomized trials are lacking, and most published studies are comparative with small case numbers or are non-comparative analyses or case reports.
The orbit is a quite limited region of the human body, encompassing numerous tissues that fall under the care of clinicians in the head and neck disciplines. When surgery is indicated, neurosurgeons, maxillofacial surgeons, oto-rhino-laryngologists, and ophthalmologists can be crucially involved. Furthermore, numerous other disciplines also have a role in good management of these diseases, including internal medicine, pediatrics, radiology, and hemato-oncology. Finally, pathologists make essential contributions to diagnosis by means of modern methods (e. g., molecular pathology).
#
3. History-taking and Diagnostics
3.1 History-taking
For conditions involving the area of the orbit, structured history-taking must always precede targeted diagnostics and therapy. It is useful to identify and assess key symptoms, quantify manifestations, and prioritize pathological changes in accordance with symptoms.
After the patient interview, leading symptoms must be retrieved and classified (by duration, intensity, or progression) (infobox 1).
Leading orbital symptoms
-
Exophthalmos/enophthalmos
-
Ptosis/eyelid retraction
-
Disorders of the eye motility/diplopia
-
Periocular/ocular reddening and swelling
-
Periocular congestion/chemosis
-
Periorbital/orbital pain
-
Foreign body sensation/epiphora
-
Retrobulbar pressure sensation
-
Bulbar movement pain
-
Bulbar repulsion pain/difficult repulsion
-
Elevated intraocular pressure
-
Choroid folds/papilledema
-
Visual field loss
-
Loss of vision
The history-taking also should include the patient’s background regarding general diseases as well as social and family history. In these cases, the focus must be placed on autoimmune diseases (e. g., autoimmune thyroid disease), other chronic inflammatory diseases (e. g., granulomatosis with polyangiitis), malignancies (e. g., prostate, breast, or bronchial cancer including existing genetic predisposition), and current medications (especially anticoagulants, among others). Known previous diseases of the orbit and neighboring structures (nose, paranasal sinuses, neurocranium) may give hints about recurrent conditions.
Because diseases of the orbit may lead to vision loss and frequently manifest primarily ophthalmologically, taking an eye-related history is of highest importance. Also important is an examination to determine if both eyes are functionally equal and to identify any amblyopia, to establish whether previous surgeries have been performed in the area of the eyes, and to try to determine how visual acuity and refraction (refractive power rates) have developed in the context of possible orbital disease. The leading symptom of “exophthalmos” might be confused with pseudo-exophthalmos (e. g., upper eyelid retraction) or enophthalmos of the contralateral side.
#
3.2 Ophthalmological diagnostics [1]
The diagnostic approach to assessing for orbit-related conditions includes the following:
-
Inspection, palpation, and measurement of the palpebral fissure, determination of the repositioning of the eyeballs, testing of corneal sensitivity and intraocular pressure;
-
Determination of vision, including refraction;
-
Perimetry assessments, including visual field examination with determination of the stimulus threshold and measurement of the visual field limits and the blind spot;
-
Exophthalmometry and orthoptics; and
-
Split lamp examination and ophthalmoscopy with assessment of the papilla.
For detailed descriptions regarding ophthalmological history-taking and examination techniques, a recent continuing medical education article covers the specific information [2].
#
3.3 Radiological diagnostics (MRI, CT, ultrasound)
Radiological procedures allow for:
-
Differential diagnostic classification,
-
Determination of extent,
-
Display of possible infiltration of surrounding structures,
-
Surgery planning (CT scan/MRI for navigation), and
-
Radiation planning.
Ultrasound can give initial hints about the type of tumoral mass but is limited regarding measurement of depth. MRI is superior to CT scan in cases of unclear inflammatory lesions and tumors. T1- and T2-weighted and T2 fat-saturated sequences (which afford excellent visualization of the optic nerve and the eye muscles) are performed. The additional application of contrast agents is suitable for examinations associated with tumors and inflammatory processes.
A CT scan allows for the best identification of calcified and bony structures with sufficient significance for soft tissue structures. Thus, this modality plays an important role in surgery planning. In cases of unclear masses and inflammation, the application of contrast agents may give additional information concerning the tissue type and vascularization. One disadvantage is radiation exposure, which can damage the eye lens and therefore should be applied sparingly in young patients.
Angiography, scintigraphy, PET, and SPECT are reserved for exceptional cases. For documentation and planning of reconstructions, photography, 3D photography, and perhaps surface scans also may be relevant [3].
#
3.4 Histopathological diagnostics
If a clinical diagnosis cannot be made based on the above-mentioned examination techniques, biopsy with histopathological examination is important and suitable, in particular in the context of unclear or advanced findings, recurrence, or therapy failure. In these cases, the risks of iatrogenic damage should not outweigh the benefit. For targeted planning of biopsy, adequate clinical examination and imaging with subsequent interdisciplinary discussion are advised. The following recommendations should be considered regarding biopsies:
-
Intake of systemic corticosteroids and other immunosuppressants should be avoided in the weeks before biopsy, if possible.
-
Incisional or excisional biopsies can be taken for formalin fixation or examinations of fresh material.
-
The biopsy should afford sufficiently large specimens, as morphological examination of fine needle aspirates is not always possible (and may be sufficient only in cases of lymphoma or metastasis).
-
Biopsy samples should be taken from different areas of a lesion.
-
Damage to the examination material should be avoided.
-
Material should be compared to previous biopsy samples of other organs, if available, for better identification of systemic diseases.
#
3.5 Laboratory diagnostics
For a multitude of rare orbital diseases, further examinations are useful, in particular adequate laboratory diagnostics. Readers interested in specific details can refer to [4].
In the following sections, we systematically illustrate the various rarely occurring structural, inflammatory, degenerative, and neoplastic changes involving the orbit.
#
#
4. Structural Lesions of the Orbit
Structural lesions of the orbit include congenital and acquired changes. The first category comprises hamartomas, choristomas, teratomas, and tissue ectopy and the large group of bone anomalies. Acquired alterations largely involve post-inflammatory and post-traumatic conditions.
4.1 Cysts and cystic lesions
Cystic lesions may occur as isolated or multiple findings. They are usually more or less spherical and can have different consistencies (e. g., serous, sebaceous, solid, mixed). In general, these lesions are painless masses that grow quite slowly. In the context of secondary inflammatory reactions, a local granulomatous tissue reaction may occur, leading to severe reactive changes and to osteolysis in the area of bone structures.
Below are some aspects that contribute to classification:
-
Timing of manifestation (congenital or acquired)
-
Number (isolated or multiple)
-
Tissue type (epithelial or non-epithelial)
-
Location
-
subperiostal
-
extraconal
-
conal
-
intraconal
-
-
Etiology
-
vascular/hematogenous
-
neurogenic
-
infectious
-
metastatic
-
4.1.1 Congenital cysts and cystic lesions of epithelial origin: dermoids [5] [6] [7] [8]
-
Epidemiology
-
3–9% of the orbital masses
-
-
Etiology
-
congenital as choristoma
-
-
Location
-
mostly extraorbital (preseptal) in a temporal-superior location, along the frontozygomatic suture (70–90%) ([Fig. 1])
-
less frequently in the area of the medial upper eyelid
-
rarely as intraorbital dermoids
-
-
Clinical
-
well-delimited cystic lesion with slow progression
-
displacement of adjacent structures
-
sonographically well-defined, anechoic, oval/round lesion with hyperreflective echoes as an indication of epidermal differentiation (adnexa structures, sebum, cellular detritus), or secondary calcifications
-
fibrous capsula
-
Cave: ruptured dermoid cysts induce granulomatous inflammation; acute and chronic courses are known; And transsutural growth is possible (sandglass configuration)
-
-
Therapy
-
complete excision with preservation of the cystic wall; proliferation-active areas (mostly near the bone) especially have to be removed
-
marsupialization
-
Cave: incomplete excisions may – although rarely – lead to recurrences; more rare are malignant transformations in the sense of squamous cell carcinomas
-
-
Important differential diagnoses
-
temporal
-
primary and secondary lacrimal gland tumors
-
-
medial
-
retention cysts and mucoceles (history regarding bone lesions)
-
meningoceles; encephaloceles
-
choristomas of the nasal mucosa
-
-
general
-
each solid, non-infiltrative mass with cystic shape
-
-
further cystic lesions are conjunctival cysts, epidermoids, and cysts originating from tissue ectopy (e. g., ectopic tissue of Rathke’s pouch), cholesterol granulomas, and cholesteatomas
-
If a cystic structure is found within the tissue (e. g., the external eye muscle), a secondary process (infection, metastasis) may be suspected.

#
4.1.2 Acquired cysts and cystic lesions
4.1.2.1 Mucoceles [9] [10] [11]
-
Epidemiology
-
3–4% of orbital masses
-
age peak in the 4th to 6th decades of life
-
-
Etiology
-
missing ventilation of the affected paranasal sinus
-
previous trauma/surgery in 50%
-
common in children with cystic fibrosis
-
-
Location
-
medial to medial-superior in the orbit, accordingly originating from the ethmoid or frontal sinus
-
rarely originating from the sphenoid sinus
-
subperiostal to diffusely intraorbital as a complication of, e. g., orbital phlegmons
-
-
Clinical
-
exophthalmos/bulbar displacement, diplopia ([Fig. 2])
-
pain only in the context of secondary inflammatory changes of the orbit; otherwise, often initially painless
-
displacing growth and secondarily compression of the optic nerve, the ocular bulb, or the draining lacrimal pathways
-
Cave: enophthalmos is a possible rare complication; possible osteolysis of the orbital floor in the context of “silent sinus syndrome” of the maxillary sinus; opticus atrophy possible with chronic course
-
-
Therapy
-
restoration of adequate ventilation of the affected sinus
-
excision of the cystic mass
-
complication management (incision, drainage, intravenous antibiotics)
-
In cases of fronto-ethmoid location, meningocele/encephalocele must be considered. Hypertelorism and increased size during Valsalva maneuver are additional indicators.
CaveBilateral fronto-ethmoid mucoceles also induce Hypertelorism.
#
4.1.2.2 Dacryops [12] [13]

-
Epidemiology
-
0.5–2% of orbital masses
-
usually in the 2nd to 5th decades of life
-
-
Etiology
-
occlusion of the glandular excretory duct (after inflammation/trauma)
-
multifactorial
-
idiopathic
-
-
Location
-
mostly palpebral part of the lacrimal gland (less frequently: orbital part, accessory lacrimal glands, cysts in the ectopic lacrimal gland tissue, or cysts of glandular tissue of the caruncle; [Fig. 3])
-
isolated, rarely multiple and bilateral occurrence
-
-
Clinical
-
more or less pronounced paragraph shape of the upper eyelid (temporal pseudoptosis)
-
foreign body sensation
-
rapidly increasing size possible in the context of emotional reflex secretion or following bleeding (color change)
-
depending on the size, astigmatism and/or diplopia
-
Cave: rarely, secondary bacterial colonization
-
-
Therapy
-
complete excision of cyst(s)
-
marsupialization, laser photocoagulation
-
wait-and-see, tear substitute, avoiding cold wind/water
-
-
Important differential diagnoses
-
rarely dacryops as sequela of trachoma, pemphigoid, or dacryolith
-
#
4.1.2.3 Orbital implantation cysts [14] [15]
-
Epidemiology
-
heterogeneous data depending on the etiology (most frequently after enucleation with an incidence of 2–7% of cases)
-
-
Etiology
-
posttraumatic (e. g., strabismus surgery, peri- and intraocular surgery, penetrating orbital trauma, enucleation, orbital fracture) because of dissemination of (conjunctival) epithelium
-
-
Clinical
-
slowly growing cystic tumor
-
Cave: rupture leading to granulomatous inflammation
-
-
Therapy
-
complete surgical excision
-
#
#
#
4.2 Neurogenic cysts
These rare anomalies are associated with congenital disorders of the eye, optic nerve, or meninges.
4.2.1 Congenital cystic eye [16] [17] [18]
-
Synonym
-
anophthalmos with orbital cyst
-
-
Epidemiology
-
quite rare
-
-
Etiology
-
consequence of non-invagination of the primary optic vesicle
-
multifactorial
-
-
Location
-
central to superior in the orbit (protruded upper eyelid)
-
-
Clinical
-
mostly isolated, rarely syndromic (Orbeli syndrome)
-
cyst, sometimes lined by neuroglia
-
missing ocular bulb
-
possible contralateral occurrence of microphthalmos
-
mostly associated with systemic pathologies
-
agenesis of corpus callosum, basal encephaloceles, heterotopia of the gray matter
-
facial clefts, saddle nose, choanal atresia, sphenoid anomalies
-
genital malformations
-
finger malformations
-
Cave: possible connection to the arachnoid space
-
-
-
Therapy
-
depending on cyst size, indication for surgical excision
-
in cases of small, constant cysts, wait-and-see is justified
-
in cases of high surgical risk, cyst puncture should be considered
-
-
Important differential diagnoses
-
teratomas with a solid component in addition to the cystic part and showing (sometimes rapid) growth
-
#
4.2.2 Microphthalmos, microphthalmos with cyst, and anophthalmos [16] [17] [18] [19] [20] [21] [22] [23] [24]
-
Epidemiology
-
anophthalmos: 1–4:100 000
-
microphthalmos: 2–20:100,000; among them 2–5% with additional cysts
-
microphthalmos with cyst in 2% of cystic orbital lesions
-
-
Etiology
-
sequela of incomplete invagination of the primary optic vesicle
-
multifactorial
-
SOX2 gene on chromosome 3 with anophthalmos; additional associated genes (e. g., PAX6, OTX2, CHS10, FOXE3, and RAX)
-
microphthalmos/anophthalmos, isolated or in the context of syndromic malformations (>30% syndromic;>57% multiple congenital malformations)
-
predisposing factors
-
pregnancy-related (vitamin A deficiency, radiation exposure)
-
infections (e. g., rubella)
-
diabetes mellitus
-
substance abuse
-
medications
-
consanguinity
-
-
-
Location
-
unilateral:bilateral ratio, 2:1 for anophthalmos
-
mostly unilateral microphthalmos
-
microphthalmos with inferior cyst and protruding lower eyelid (compare delimitation of cystic eye)
-
-
Classification of microphthalmos/anophthalmos ([Table 1], infobox 2)
-
unilateral vs. bilateral
-
with or without other ocular pathologies
-
with or without systemic manifestation
-
syndromic or non-syndromic
-
-
Clinical
-
severe malformations and functional blindness of the contralateral eye in 12.5% of unilateral microphthalmos cases and 34.0% of unilateral anophthalmos cases
-
cerebral pathologies, mainly malformations in the area of the corpus callosum, occurring in cases of bilateral anophthalmos (70%), unilateral anophthalmos (20%), and unilateral microphthalmos (12.5%)
-
association with clefts
-
possible association with different internal diseases (cardiovascular, pulmonary, renal, gastrointestinal)
-
rare associations
-
Waardenburg syndrome (inner ear hearing loss)
-
congenital ectodermal dysplasia
-
-
-
Therapy
-
indication for surgical excision depending on cyst size
-
in cases of small, stable cysts, wait-and-see is justified
-
in cases of high surgical risk, cyst puncture possible
-
enlargement of the hypoplastic orbit with volume expanders
-
-
Important differential diagnoses
-
teratomas, cryptophthalmos (infobox 3) and bulbar phthisis as degenerative (post-inflammatory) alteration (bulbus quadratus)
-
A continuum has been described, from sighted, severely hyperopic microphthalmos to anophthalmos. The orbit with adnexa structures (eyelids and extraocular muscles) may show near-normal development. Bulbar rudiments that are in part microscopically small can be present so that true anophthalmos can be extremely difficult to define. Some authors diagnose (true, complete) anophthalmos only when histological examination of the orbital contents confirms the absence of ocular tissue. This gray area is why the term ‘clinical anophthalmos’ or ‘incomplete anophthalmos’ was coined
|
Term |
Characteristics |
|---|---|
|
Simple microphthalmos (synonym, nanophthalmos) |
|
|
Microphthalmos with coloboma, possibly with cyst |
|
|
Microphthalmos without confirmed coloboma |
|
|
Microphthalmos with systemic manifestation |
|
|
Further occurrence of microphthalmos: dyscephalia oculo-mandibulofacial Ullrich-Fremery-Dohna, type Francois, dyscephalia according the Hallermann-Streiff, dysplasia oculo-vertebralis van der Hoeve syndrome, Potter syndrome, Fanconi syndrome. |
Anophthalmos
-
Complete absence of ocular tissue
-
primary anophthalmos – development of neither eye nor orbit
-
secondary anophthalmos – non-viable malformations in combination with cranial malformations
-
degenerative anophthalmos – ocular primordium present but degenerated
-
-
Frequently hypoplastic and thus size-reduced orbit but with enormous self-differentiation capacity
-
agenesis
-
complete anophthalmos, also secondarily possible
-
presence of mesodermal tissue
-
presence of ectodermal tissue
-
presence of meso- and ectodermal tissue
-
-
Possibly involving malformations of the visual pathway and agenesis of the corpus callosum
-
Malformations of the skull, meningoceles, orbital dermoids, deafness
Cryptophthalmos
-
Absence of eyelids in combination with microphthalmos/anophthalmos
-
Hypoplastic orbit
-
Eyebrow hairiness incomplete or completely missing
-
Mostly bilateral
-
In cases of unilateral occurrence, severe malformations of the orbit and of the contralateral eye
-
Orbital roof absent, malformation of the sphenoid bone, auricular anomalies, syndactyly, facial clefts, laryngeal atresia, anal atresia, genital malformations, meningo-encephaloceles, hydrocephalus, persistent craniopharyngeal duct (orbital cyst formation from residues of Rathke’s pouch), pituitary gland disorders, adrenal aplasia
#
4.2.3 Orbital cephaloceles [16] [25] [26]
-
Epidemiology
-
cephaloceles: overall 0.8–5:10,000; occipital in>70% of cases
-
overall<1% of orbital masses in children
-
-
Etiology
-
deficient separations of neuroectoderm and ectoderm
-
persisting connections between the neurocranium and the orbit
-
-
Classification
-
anterior (frontal bone, ethmoid bone, lacrimal bone, maxilla)
-
posterior (sphenoid bone)
-
-
Clinical
-
hypertelorism, pulsatile exophthalmos
-
painless (pulsatile) tumor
-
frontal sinus aplasia
-
rarely bilateral, but if so, then associated with hypertelorism
-
differentiation between meningoceles (meninges) and meningoencephaloceles (meninges and brain tissue) depending on contents
-
further skull anomalies possible (sphenoid), hydrocephalus
-
association with neurofibromatosis
-
Cave: possible secondary ulceration and infection of the cephaloceles
-
-
Therapy
-
excision and closure of the bone defect
-
duraplasty
-
Generally, these lesions are present at birth but sometimes will manifest only later in life (especially when located dorsally).
#
4.2.4 Other neurogenic cysts [27]
Primary arachnoid cysts may be found in combination with ipsilateral ocular colobomas. Nerve sheath cysts as a further entity are associated with other anomalies of the central nervous system (CNS).
#
#
4.3 Congenital tumors and tissue ectopy
4.3.1 Dermolipoma [28]
-
Epidemiology
-
about 2% of orbital tumors in children
-
-
Etiology
-
choristoma
-
ectopic ectoderm disseminated as epidermal tissue of the eyelids in the area of the conjunctiva and frequently in an intraorbital direction
-
-
Location
-
temporal palpebral area
-
rarely along the inferior and superior conjunctival fornix
-
-
Clinical
-
often asymptomatic
-
possibly keratinizing
-
partly bearing hair follicles
-
foreign body sensation
-
-
Therapy
-
surgical excision with preservation of the conjunctiva, lacrimal gland, and exterior ocular muscles (lateral rectus muscle) if desired (esthetic) or in cases of irritation of the ocular surface
-
superficial removal without preparation into the depth of the orbit is sufficient in most cases
-
-
Important differential diagnoses
-
lymphoma
-
orbital fat hernia ([Fig. 4])
-

#
4.3.2 Ectopic lacrimal gland tissue and other tissue ectopy [29] [30]
Accessory lacrimal gland tissue is regularly found in the area of the conjunctival fornices (Wolfring’s and Krause’s glands). In the posterior areas of the orbit, this tissue is rarely found. However, it may induce chronic scarring inflammation where it occurs that requires surgical intervention.
In addition, heterotopic cranial tissue and cartilage structures have been described in the orbit.
#
4.3.3 Orbital teratoma [31] [32] [33]
-
Epidemiology
-
6.6% of pediatric tumors, mostly located outside the orbit
-
quite rare orbital occurrence
-
-
Etiology
-
neoplasm of two or all three germ layers (pluri- to totipotent embryonic stem cells)
-
-
Clinical
-
progressive, unilateral, mostly cystic tumor
-
proptosis
-
possible enlargement of the bony orbit by a factor 2 or 3
-
strongly heterogeneous tissue
-
complete teratoma as orbital fetus-in-fetu (orbitopagus parasiticus)
-
incomplete second fetus with parts of a spinal cord
-
teratoma with evidence of all three germ layers
-
dermoid tumor with evidence of two germ layers
-
-
-
Therapy
-
surgical excision
-
-
Important differential diagnoses
-
dermoid, rhabdomyosarcomas, or vascular anomalies
-
cave:
-
malignant teratoma of the orbit (in 2% of cases)
-
intracranial extension
-
-
#
#
4.4 Bone anomalies and mesodermal defects
4.4.1 Accessory bones and sutures, osseous variations, aberrant foramina [34] [35] [36]
-
Epidemiology
-
some types are frequent (duplication of the fronto-zygomatic suture in 7 of 400 skulls)
-
significant regional differences
-
found in about 1–2% of the orbits
-
-
Etiology
-
disturbed ossification
-
classification into variations and congenital deformities
-
orbit mostly self-determined regarding development (independently from the eye)
-
anatomical pathways are only secondarily surrounded by bone; nerve and vessel duplications precede bone variations
-
-
Clinical
-
partly clinically completely unapparent
-
association with dysostoses of the skull
-
exophthalmos
-
facial asymmetry
-
hyper- or hypotelorism
-
4.4.1.1 Examples of bone variations [34]
-
Accessory sutures and bone fragments (e. g., duplication of the fronto-zygomatic suture, fragmented zygomatic bone with ossiculum infraorbital marginale)
-
Missing involvement of the maxilla in the development of the inferior orbital margin (incidence of 1:2250)
-
Malformations of the ethmoid bone where the frontal bone forms the medial orbital wall
-
Frequent dehiscence of the lamina papyracea
-
Incomplete presence up to complete absence of the lacrimal bone; variations of the lacrimal hamulus and thus variations of the lacrimal sac and nasolacrimal duct
-
Accessory ossicles in the area of the frontal maxillary process
-
Maxillary hypoplasia
-
Duplication of the anterior lacrimal crest
#
4.4.1.2 Examples of foramina variations [34]
-
Supra- and infraorbital foramina in form of channels, incisions/sulcus, multiple primordia
-
Duplication of the optic canal/missing optic foramen in cases of anophthalmos
-
Up to four-fold primordium of the zygomatico-facial foramen
-
Up to five-fold primordium of the infraorbital foramen (double primordium in>10%)
-
Trochlear spine (simple, double, or as ring)
-
Bony separation of the inferior orbital fissure by an accessory bone bridge
#
4.4.1.3 Examples of wall defects [34]
-
Unilaterally missing development of the frontal bone and the maxilla (frontal lobe covered only by meninges and skin; clinically also without presence of cephaloceles with downward displacement of the eyeball and divergence)
-
Cave: association with cyclopia and arhinencephaly, rare coincidence with phacomatoses, e. g., in cases of neurofibromatosis (malformations in the area of the sphenoid bone)
#
4.4.1.4 Examples of wall dehiscence [34]
-
Infantile and sometimes senile physiological occurrence
-
Especially in the area of the maxilla and the ethmoid bone
-
More rarely in the area of the lacrimal bone, palatine bone, or sphenoid bone
-
Cave:
-
optic canal variation because of absent parts of bone surrounding the optic nerve and the resulting direct communication between the dural sheath and sphenoid mucosa
-
association of orbital varices with bone dehiscence or misinterpretation of phleboliths
-
#
#
4.4.2 Cribra orbitalia [34] [35]
Thinning of the orbital roof with exposed diploe and contained venous plexus because of a bone development disorder (differential diagnosis of vitamin D deficiency, anemia).
#
4.4.3 Familial hypoplasia of the orbital margin (Urrets-Zavalia syndrome) [34]
-
Epidemiology
-
extremely rare (<1:1 000 000; described in two families to date)
-
-
Etiology
-
genetic agenesis of the orbital margin and parts of the ocular adnexa structures
-
dominant inheritance (high penetration, constant expression)
-
involves tissue of the paraxial and visceral mesoderm
-
-
Clinical
-
missing bony orbital frame
-
hypoplasia of the eyelid skin and palpebral tarsus
-
variable defects with disorders of the draining nasolacrimal pathways, eyelid coloboma, vertical strabismus
-
Accessory bones and sutures or aberrant foramina often have no pathological significance but sometimes must be considered in the context of sinus, orbital, and lacrimal duct surgery.
#
4.4.4 Facial clefts, facial dystrophy, craniofacial dysostoses, and malformations of the skull [34] [37] [38]
Complex facial deformities originating from a disorder of embryogenesis in the area of the first and second pharyngeal arches.
4.4.4.1 Rare facial clefts (including Tessier clefts) [37] [38] [39]
-
Epidemiology
-
1–5:100 000
-
-
Etiology
-
lack of fusion of embryonic tissues of the first pharyngeal arch (association with malleus and incus malformations)
-
early developmental stage (embryonic weeks 5–8)
-
in later stages because of umbilical cord trauma (atypical cleft, amniotic band syndrome)
-
-
Typical type
-
oro-ocular or oro-orbital facial cleft
-
transverse and diagonal clefts
-
clinical
-
numerous manifestations
-
partly medial eyelid colobomas
-
-
-
Atypical type
-
oro-temporal facial cleft
-
clinical
-
temporal eyelid colobomas
-
association with dermoids, ear adnexae, other facial deformities (cleft lip and palate)
-
anophthalmos, microphthalmos, uveal coloboma
-
-

-
15 types (0–14), possible course from the maxilla via the orbit to the frontal bone
-
Groups
-
midline clefts
-
paramedian clefts
-
orbital clefts
-
lateral clefts ([Fig. 5])
-
-
Therapy
-
plastic reconstruction by maxillofacial surgeons to close the soft tissue and bone defect, orthodontic treatment
-
phoniatrics/pedaudiological involvement
-
if needed, oculoplastic intervention for eyelid correction or coloboma closure
-
#
4.4.4.2 Facial dystrophy, craniofacial microsomia [37] [41]
-
Epidemiology
-
1:3000 to 1:5000
-
-
Etiology
-
disturbed embryogenesis (concerning the first pharyngeal arch)
-
-
Clinical
-
jaw bones
-
asymmetric midfacial hypoplasia
-
ankyloses
-
micrognathia
-
-
eye
-
dermoid (epibulbar)
-
upward or downward displacement of the globe
-
anophthalmos/microphthalmos
-
eyelid coloboma
-
-
mouth
-
macrostomy (clefts)
-
facial clefts
-
-
skeletal system
-
changes in the spine
-
-
cranial nerves
-
facial nerve palsy
-
sensory hearing loss
-
palatal lift disorder
-
ocular muscle paresis
-
-
ear
-
ear adnexae
-
microtia/anotia
-
auricular canal atresia
-
conductive hearing loss
-
-
Cave: rare association with tetralogy of Fallot, ventricular septum defect, transposition of the major vessels, aortic arch anomalies, kidney malformations (absence of one kidney, duplicate ureter, renal ectopy), hydronephrosis, hydroureter, anomalies of the extremities (ulna, radius), microcephaly, encephalocele, hydrocephaly, corpus callosum hypoplasia, Arnold-Chiari malformation, holoencephaly
-
present as syndromes: VA(C)TER(L) – vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal atresia, renal anomalies, and limb anomalies; or CHARGE – coloboma, heart, atresia choanae, retardation of growth and development and genitourinary and ear anomalies
-
-
Types ([Table 2])
-
otocephaly
-
mandibulo-facial dysostosis
-
oto-mandibular dysostosis
-
oculo-auricular dysplasia
-
mandibulo-oculo-facial dyscephaly
-
oculo-vertebral dysplasia
-
microgenia and glossoptosis
-
|
Term |
Described by |
Laterality |
Characteristics |
|---|---|---|---|
|
Otocephaly |
St. Hilaire |
Bilateral |
|
|
Mandibulo-facial dysostosis |
Franceschetti, Zwahlen, Klein, Treacher-Collins |
Bilateral |
|
|
Oto-mandibular dysostosis |
Francois and Haustrate |
Unilateral |
|
|
Oculo-auricular dysplasia |
Goldenhar |
Unilateral |
|
|
Mandibulo-oculo-facial dyscephaly |
Hallermann and Streiff, Ullrich and Fremerey-Dohna |
Unilateral |
|
|
Oculo-vertebral dysplasia |
Weyers and Thier |
Unilateral |
|
|
Microgenia and glossoptosis |
Pierre Robin |
|
#
4.4.4.3 Facial (hemi)atrophy (Parry-Romberg) [42] [43]
-
Synonym
-
progressive facial trophoneurosis
-
-
Epidemiology
-
1:700 000
-
females more frequently affected than males
-
-
Etiology
-
uni- or rarely bilateral disorder of the soft tissue and bone because of missing/insufficient innervation (cranial nerves V and VII)
-
-
Clinical
-
slowly progressive, generally unilateral atrophy of the facial soft tissue including muscles as well as some of the bony-cartilaginous structures
-
facial asymmetry
-
highly variable disease activity over 2 to 20 years until self-limiting stagnation
-
malformation of the external auricle (misplaced ear, auditory canal atresia, adnexae, reduced ossicles, small tympanum)
-
unilateral micrognathia
-
hypoplastic mastoid
-
atrophic facial muscles, yellowish skin (possible hypo- and hyperpigmentation), lanugo hairiness with otherwise alopecia
-
temporally sloping axis of the eyelid, enophthalmos, lagophthalmos, pseudoptosis (differential diagnosis of associated Horner’s syndrome)
-
exposed facial nerve in the middle ear
-
atrophy of soft palate and tongue (speaking and swallowing disorders)
-
saber-like scleroderma along the medial orbital skin
-
heterochromia, blepharophimosis, coloboma, oculo-motor paresis, partly nystagmus
-
Cave:
-
rare hemiatrophy of the entire body
-
frequent occurrence of orbital tumors (among others neurinomas)
-
-
-
Therapy
-
systemic medication (including glucocorticoids, cyclophosphamides, methotrexate) and local therapies (among others PUVA, botulinum toxin, phototherapy)
-
surgical therapy options for stabilization or reconstruction in cases of periocular changes
-
-
Important differential diagnoses
-
facial hemihypertrophy
-
unilateral hypertrophy of one side of the body
-
exophthalmos with megalophthalmos
-
-
congenital facial nerve palsy (Möbius syndrome)
-
#
4.4.4.4 Craniofacial dysostoses [44] [45] [46] [47] [48]
Extreme heterogeneity of diseases with different nomenclature
-
Epidemiology
-
syndromic and non-syndromic
-
3–10:10,000
-
syndromic types occur 10–50 times more rarely; extremely rare types<1:1,000,000 (e. g., Boston syndrome)
-
most frequent syndromes
-
Crouzon syndrome
-
Pfeiffer syndrome
-
Saethre-Chotzen syndrome
-
-
rare syndromes
-
Boston syndrome
-
Cole-Carpenter syndrome
-
Herrmann-Opitz craniosynostosis
-
craniosynostosis, Philadelphia type
-
cardio-cranial syndrome, Pfeiffer type
-
Jackson-Weiss syndrome
-
Hunter-McAlpine craniosynostosis
-
Lopez-Hernandez syndrome
-
Baller-Gerold syndrome
-
-
-
Etiology
-
primordial disorder of the bone, premature closure of affected sutures
-
increased volume expansion in one direction because of expansive cranial growth and blockage in the other direction (Virchow’s rule: blockage perpendicular to the synostosis and compensatory growth along the synostosis)
-
metopic suture (physiologically open up to the 8th month of life), sagittal suture, lambdoid suture, coronal suture not open until adulthood
-
manifestation of the disease typically in the 1st to 4th years of life
-
-
Clinical
-
skull deformity with or without orbital involvement
-
exophthalmos, hyper- and hypotelorism, strabismus
-
increased intracranial pressure (headaches, papillary edema, opticus atrophy)
-
encephalocele
-
syndactyly
-
ear malformations (auricle, external auditory canal, middle ear)
-
association with metabolic disorders (hyperthyroidism, vitamin D–resistant hypophosphatemia, mucopolysaccharidosis [associated with dysmorphia of the head, formerly called gargoylism], and mucolipidosis), chondro- and osteodystrophies (e. g., achondroplasia)
-
4.4.4.4.1 Cleidocranial dysostosis [44] [49]

-
Hypoplastic dysostosis of the skull with brachy- or platycephaly ([Fig. 6])
-
Dental anomalies
-
Pseudoarthrosis, hypoplasia or aplasia of one or both clavicles
-
Hypoplasia of the nasal bone and maxilla
-
Elevated palate
-
Possible deformities of thorax, pelvis, and extremities
-
Spina bifida
#
4.4.4.2 Oxycephaly (acrocephaly, tower skull) [44] [45] [46] [47] [48]
-
Male:female ratio, 4:1
-
Vertical elongation of the skull
-
Transverse and sagittal shortening
-
Coronary and facial synostoses
-
Reduction of the skull base
-
Prominent nose, hypoplastic maxilla, narrow palate
-
Flat orbits with small volume
-
Supraorbital pronounced exophthalmos, strabismus, motility disorders, nystagmus, exposition keratopathy, mild hypertelorism
-
Papillary edema, opticus atrophy
-
Retinal vascular stasis, tortuositas vasorum
-
Eyelid edema
-
Rare associations: cataract, corneal dystrophy, eyelid coloboma, orbital encephaloceles
-
Often minor impairment of intelligence (Cave: apparent intellectual disability often possibly a consequence of psychosocial stigmatization and not biological)
-
Classic syndrome: Apert syndrome (acrocephalo-syndactyly)
-
Oxycephaly with exophthalmos, exposition keratopathy, ophthalmoplegia, papillary edema, opticus atrophy
-
Syndactyly (second to fourth fingers/toes); synarthroses (shoulder, elbow)
#
4.4.4.3 Scaphocephaly (sphenocephaly) [44] [45] [46] [47] [48]
-
Synostosis of the sagittal suture
-
Long small shape of the head
-
Low width of the skull with hypotelorism
-
Sagittal synostosis
#
4.4.4.4 Brachycephaly [44] [45] [46] [47] [48]
-
Synostosis of the coronal or lambdoid sutures
-
Short, broad skull
-
Intermediate between scaphocephaly and oxycephaly
#
4.4.4.5 Dolichocephaly [44] [45] [46] [47] [48]
-
Prominent frontal skull
-
Coronal, sagittal, and/or lambdoid synostoses
-
Coronal representative: Crouzon syndrome
-
prominent front, typical shape of the nose (parrot beak)
-
hypoplastic maxilla, prognathism, malpositioned teeth, elevated palate, partly with cleft formation
-
diverging strabismus, lacrimal duct stenosis, papillary edema, opticus atrophy, exposition keratopathy, ocular coloboma, glaucoma, ectopia lentis
-
ear anomaly (auditory canal atresia, auricular anomaly)
-
#
4.4.4.4.6 Plagiocephaly [44] [45] [46] [47] [48]
-
Sagittal asymmetry
-
Unilateral synostosis of different sutures
-
Classified into:
-
anterior type
-
unilateral synostosis of the coronal suture
-
hypoplastic ipsilateral orbit
-
orbital roof with declining axis in temporal direction
-
-
posterior type
-
closure of the lambdoid suture
-
to be delimited against acquired positional plagiocephalus by differential diagnosis
-
-
#
4.4.4.4.7 Hemicraniosis [44] [45] [46] [47] [48]
-
Prominent frontal bone, parietal bone, and zygomatic bone
-
Significant asymmetry
-
Exophthalmos and opticus atrophy
#
4.4.4.4.8 Trigonocephaly [44] [45] [46] [47] [48]
-
Frontal cranial dysplasia (synostosis of the metopic suture) with occipital compensation
-
Also with coronary synostosis
-
Hypotelorism
-
Opticus atrophy
-
Narrow anterior cranial fossa
-
Hypoplastic ethmoid sinus
#
4.4.4.4.9 Platycephaly (clinocephaly) [44] [45] [46] [47] [48]
-
Increased posterior cranial fossa with overriding of the cervical spine
-
Synostosis of the superior sutures
-
Therapy:
-
conservative by changing the skull position, i. e., through physiotherapy or helmet wearing
-
correction or prevention of increased intracranial pressure
-
treatment of the head deformity (functional/psychosocial indication)
-
surgery techniques:
-
open or minimally invasive suturectomy
-
suturectomy and removal or remodeling of single bone fragments, if needed as fronto-orbital advancement
-
cranial distraction
-
-
#
#
4.4.4.5 Hypertelorism [37] [44] [45]
-
Interorbital distance of>30 mm
-
Association with craniosynostosis or cleft formation in the maxillary and facial area, but also isolated
-
Rare occurrence of frontal or frontobasal encephaloceles
-
Classification
-
primary
-
morphogenetic (habitual) because of disproportional growth
-
embryonic because of developmental disorders (e. g., Apert and Crouzon syndrome) with nasal cleft (rarely duplicate nasal septum, dermoid)
-
-
secondary
-
developmental disorder attributable to environmental factors (trauma, encephaloceles, diseases of the cartilage or bone)
-
-
#
4.4.4.6 Median cleft face syndrome [50]
-
Synonym/manifestation
-
cyclopia, holoprosencephaly, arhinencephaly, otocephalic anomalies
-
-
Epidemiology
-
1:16 000 live births
-
-
Etiology
-
complex cerebral malformation with missing hemisphere separation
-
cyclopia from disturbed balance of excitation and inhibition processes of development and concerning the entire cranial position
-
-
Clinical
-
sometimes presence of proboscis as rudiment of the nasal apparatus
-
malformations such as completely missing eyes (anophthalmos) or face (aprosposus, i. e., missing eyes and olfactory and gustatory organs) as well as presence of one (cyclopia or synophthalmia) via three (diprosopus triophthalmus, cephalotheracopagus) or four separated eyes (tetrophthalmus) possible
-
starting at the optic nerve, possible separated presentation of both eyes and orbits from posterior in anterior direction (for example, a single cornea with otherwise duplicated tissues)
-
many clinical variations with fluent transition, e. g., median facial clefts, microphthalmos, anophthalmos, cyclopia, hypotelorism (facial dysmorphia correlating with cerebral anomaly)
-
isolated or in the context of syndromes (including CHARGE, Smith-Lemli-Opitz syndrome, Rubinstein-Taybi syndrome, Meckel syndrome, Lambotte syndrome, Steinfeld syndrome)
-
complications
-
neurological: epilepsy, hydrocephalus, developmental intellectual disability, hypotonia, spasms
-
craniofacial: microcephaly, hypo- or hypertelorism, median and lateral clefts (concerning lips, maxilla, and palate), arhinia, hypoplasia of the piriform aperture
-
endocrinological: diabetes insipidus, growth hormone deficiency, hypoplasia of the adrenal cortex, hypogonadism
-
otomotor dysfunction: speaking and swallowing disorders, gastroesophageal reflux, necessity for gastrostomy
-
vegetative dysfunctions: temperature regulation, heart and respiratory rate control
-
-
life expectancy
-
unfavorable prognosis, lethal in early childhood (mean mortality at age 4 years; 15% between ages 10 and 19 years)
-
-
-
Therapy
-
symptom-based
-
#
#
4.4.5 Developmental disorders of the bones
4.4.5.1 Fibrous dysplasia [51]
-
Etiology
-
mesenchymal defect of the bone spongiosa and the bone marrow and replacement by fibrous tissue
-
-
Clinical
-
subtypes:
-
affecting only one bone (monostotic)
-
affecting several bones (polyostotic)
-
McCune-Albright syndrome (severest type of microsomia, endocrine and pigment disorders of the skin)
-
-
possibly affecting skull
-
in cases of cranial location, orbital involvement in>40%
-
atypical facial pain and headache
-
sinusitis
-
hearing disorders
-
exophthalmos
-
facial asymmetry
-
-
Therapy
-
surgical decompression (optic channel, orbit) in cases of clinical complaints such as optic neuropathy
-
surgical excision of involved areas with consideration of functional aspects (curative surgery is not possible; basically, a benign disease)
-
Cave: radiation therapy contraindicated because of the high risk of malignant transformation
-
#
4.4.5.2 Osteopetrosis [52]
-
Epidemiology
-
1:250 000 to 5:100 000 (dominant inheritance)
-
-
Etiology
-
disturbed differentiation of osteoclasts
-
increasing bone substance with reduced stability
-
-
Clinical
-
macrocephaly, craniofacial dysmorphia
-
fractures, microsomia
-
nerve compressions (blindness, deafness, facial palsy)
-
osteomyelitis
-
hydrocephalus, pituitary hypoplasia, cerebral demyelination
-
choanal atresia
-
dental anomalies, double tooth series, palatal cleft, caries
-
disturbed calcium balance, secondary hyperparathyroidism
-
retinal atrophy, cataract
-
disorder of the hematopoiesis with risk of pancytopenia, hepatosplenomegaly
-
manifestations; types:
-
autosomal recessive osteopetrosis (ARO) (malignant type)
-
neuropathic type of ARO
-
association with renal tubular acidosis (i. e., RTA)
-
X-linked osteopetrosis with lymph edema, anhidrotic ectodermal dystrophy, and immune deficiency (i. e., OLE-DAID)
-
common variable immune deficiency (i. e., CVID)
-
leukocyte adhesion deficiency syndrome (i. e., LAD-III)
-
autosomal dominant type (Albers-Schönberg disease)
-
pyknodysostosis
-
dysosteosclerosis
-
osteopoikilosis (with occurrence of connective tissue naevi as Buschke-Ollendorff syndrome)
-
osteopathia striata (with or without cranial sclerosis)
-
-
-
Therapy
-
symptomatic, depending on the manifestation of the functional disorder or complication (e. g., surgical decompression of the optic nerve)
-
multi- and interdisciplinary approach
-
vitamin D substitution
-
transfusion if needed
-
#
4.4.5.3 Cherubism [53]
-
Etiology
-
different inheritance pathways, probably mainly dominant with high penetration
-
cystic proliferation of the mandible and maxilla
-
-
Clinical
-
disease onset in early childhood (2nd to 5th years of life)
-
various courses, sometimes clinically almost undetectable
-
prominent chin and cheeks, irregular tooth position
-
eyes displaced in upward direction, proptosis, diplopia
-
coarse bone swellings
-
occasional association with regional lymphadenopathy
-
spontaneous involution after bone growth (at about age 30 years)
-
Cave: dysarthria, dysphagia, and dyspnea possible
-
association with Ramon syndrome, neurofibromatosis type 1, and fragile X syndrome
-
-
Therapy
-
surgical resection of the affected region under functional aspects
-
Cave:
-
possible activation of disease by surgery
-
radiotherapy contraindicated (osteoradionecrosis, malignancy induction)
-
-
Cleft formation and other craniofacial malformations show a broad variation. They may appear as isolated or as syndromic findings. Clinically, the courses are sometimes highly complex with vital risks. The early detection and treatment of these complication-associated conditions are the major challenge in this group of diseases. The interdisciplinary approach is multimodal; often several surgical interventions are necessary.
#
#
#
4.5 Conclusion
In cases of structural anomalies of the orbit, orbital bone malformations have to be delimited from ocular pathologies. The first category encompasses mainly craniosynostoses that may occur as isolated findings or in the context of syndromes. Ocular malformations may be associated with orbital lesions (e. g., Goldenhar syndrome) or lead to them. The development of the orbit can occur completely independently from ocular organogenesis. Dermoids are the most frequently observed congenital lesions of the orbit. The major challenges are complications that require the attention of members of the neurological and/or neurosurgical disciplines (e. g., encephaloceles, hydrocephalus).
The acquired structural anomalies include mainly posttraumatic changes or sequelae of surgeries and/or radiation. These changes are highly variable and occur rather frequently.
Knowledge of possible structural changes is essential for successful therapy. The observation of the close neighborhood of the orbit to the skull is extremely important. Regarding diagnostics, appropriate imaging procedures for further topographic assessment are essential. For therapy, often an interdisciplinary exchange with neuro- and ophthalmic surgeons is necessary, initiated by the primarily treating discipline (usually ENT/maxillofacial surgery).
#
#
5. Inflammatory Diseases of the Orbit
There are many inflammatory lesions of the orbit. Below, an overview is given with classification into infectious and non-infectious types.
5.1 Infectious orbital inflammation
Infectious cellulitis is the most frequent cause of orbital inflammation. It develops mainly from inflammatory processes of the paranasal sinuses, face, oropharynx, and ocular adnexa structures, but sometimes can arise because of foreign bodies or septic dissemination. The causes are a large spectrum of bacterial and viral pathogens, fungi, and parasites.
5.1.1 Bacterial inflammation (orbital cellulitis)
The most frequent origins of infectious orbital inflammation are orbital complications of purulent sinusitis, which will not be dealt with in this article on rare orbital diseases. For reviews of their classification, pathogenesis, diagnostics, and management, see [54] [55] [56] [57] [58] [59] [60] [61] [62] [63] [64] [65] [66] [67].
The identification of rare pathogens may be a challenge but is crucial for affected patients because the diseases can entail a high local and systemic morbidity, independent of their rarity. Opportunistic infections must be considered especially in immunocompromised and previously affected patients [58]. The keys to successful management are a high index of suspicion, prompt diagnosis, and adequate therapy of the underlying disease.
5.1.1.1 Tuberculosis
In recent years, the incidence of tuberculosis has increased because of resistant strains and the distribution of immunosuppressant agents.
-
Pathogenesis: in cases of tuberculosis, the involvement of the orbit may occur via two different pathways:
-
hematogenic dissemination with two manifestation types in the orbit:
-
periostitis (more frequent): slow course with cold abscess, sequesters, and fistulas; mainly young patients affected; mostly located in the zygomatic bone
-
tuberculoma (rarer): infiltrative orbital mass, sometimes associated with neurosensory deficit
-
-
direct spread from contagious neighboring structures (paranasal sinuses or lacrimal gland); causes necrotizing infiltrative lesions, partly with cutaneous fistulas [60]
-
-
Clinical
-
painful movement impairment, no intraocular lesions, rather non-specific symptoms leading to late diagnosis
-
very rarely also atypical mycobacteria (mycobacterium hominis; mycobacterium avium)
-
Cave: especially in cases of cold abscess, wet cavities or (very rarely) complicated sphenoid osteomyelitis because of optic neuropathy, clinical suspicion often more important than blood culture for diagnosis [58]
-
-
Diagnostics
-
Therapy
-
timely tuberculostatic triple therapy (isoniazid, rifampicin, ethambutol) by specialists (not only after evidence of positive culture; the associated constellation of clinical and molecular pathological findings is crucial)
-
indication of surgical intervention for symptom relief, e. g., in cases of vision loss [58]
-
in cases of interaction between HIV and tuberculosis, sometimes complex management [60]
-
#
5.1.1.2 Syphilis
This venereal disease, which currently affects mainly immunosuppressed patients and men who have sex with men, is caused by Treponema pallidum and can involve the orbit because of post-primary hematogenic dissemination. In the orbit, it manifests as painful periostitis or a soft tissue lesion after latency [61].
-
Clinical
-
in cases of posterior manifestation, painful apex orbitae syndrome
-
focal or diffuse syphilitic periostitis into the orbital bone
-
occurrence of intraconal soft tissue lesions, in the extraocular muscles, or the lacrimal gland
-
-
Diagnostics
-
serological confirmation of the diagnosis by means of a fluorescence Treponema antibody absorption test
-
-
Therapy
-
complete healing with antibiotic therapy with penicillin G [61]
-
#
5.1.1.3 Necrotizing fasciitis
Necrotizing fasciitis of the orbit is an ophthalmological emergency but quite rare because of the good blood supply. Because of rapid progression, it can lead to death when the diagnosis is made too late [58].

-
Epidemiology
-
incidence of 0.24:100 000 [62]
-
-
Classification [63]
-
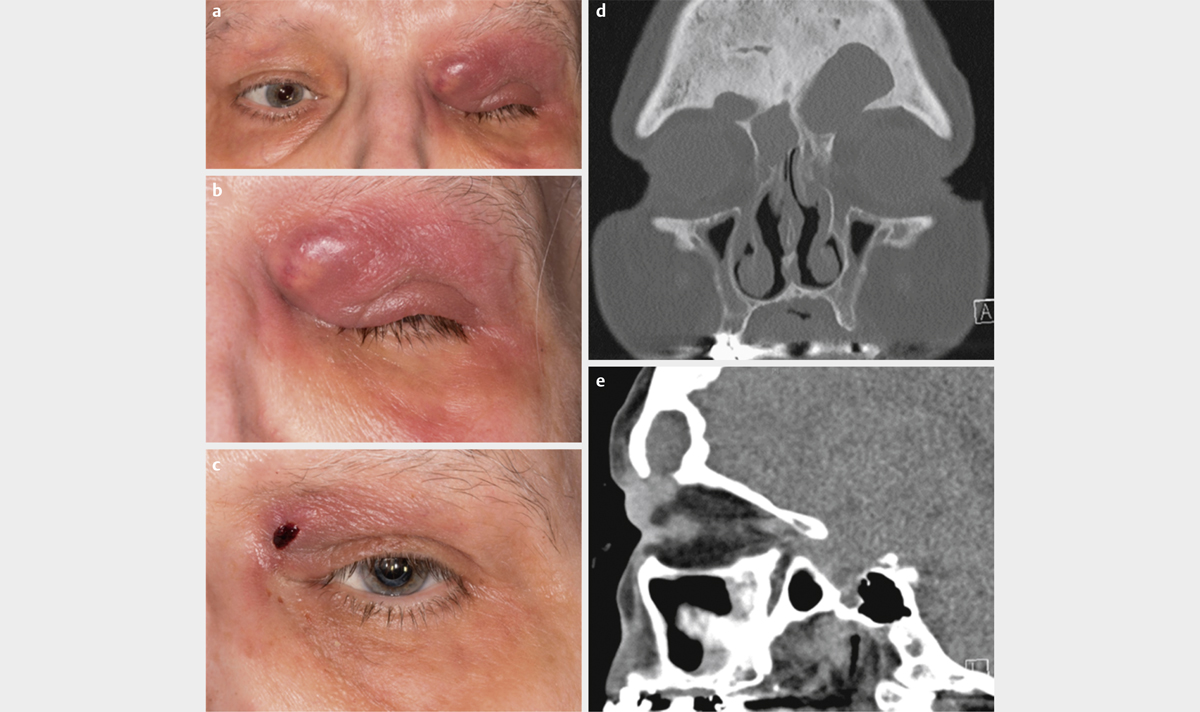
type I: polymicrobial, e. g., aerobic and anaerobic bacteria (especially in patients with multiple morbidities) ([Fig. 7a–d])
-
type II: at about 80%, the most frequent type [64] (single bacterial pathogens, mostly beta-hemolyzing streptococci of group A; superantigens and exotoxins as most important virulent factors)
-
type III: mainly vibrio bacteria; rarely occurring in Europe
-
type IV: fungi
-
-
Etiology
-
penetrating traumas are the most frequent triggers (35%), along with surgeries as well as acute infections of the paranasal sinuses, efferent lacrimal ducts, or skin, and pharmaceutical immune suppression or malignancies
-
unknown trigger in 25% of cases [64]
-
-
Risk factors
-
greater age, alcohol and drug abuse, diabetes mellitus, collagenosis, and cardiovascular disease
-
in 50% of cases, no risk factors present [64]
-
-
Clinical
-
early detection possible because of thin skin near the eye
-
severe pain possible even prior to skin changes (“pain out of proportion”) [62]
-
fever and intensive sweating
-
gangrene development within a few hours with livid, blistering skin discoloration
-
risk of blindness in cases of central artery closure [58]
-
toxic shock syndrome in 30% of cases
-
-
Diagnostics
-
based on clinical symptoms
-
scores, e. g., LRINEC [65], imaging for rapid diagnosis and delimitation of other infections such as mucormycosis possibly helpful
-
-
Therapy
-
intensive debridement (mainly subcutaneous near the eye, sometimes repeatedly performed [66] up to exenteration as ultima ratio [67]) in combination with broad spectrum antibiotics (mostly broad-spectrum penicillin or cephalosporins plus clindamycin/vancomycin; see 2009 DGHNO consensus paper [68])
-
exclusively additive effect of hyperbaric oxygen therapy and immunoglobulins [63]
-
-
Prognosis
-
mortality varies between 8 and 14%, especially high in cases of systemic complications such as toxic shock syndrome with multi-organ involvement, periorbital necrotizing fasciitis with blindness, or involvement of facial soft tissue [64] [66]
-
reduction in mortality possible through high index of suspicion and immediate therapy [58]
-
#
5.1.1.4 Non-infectious (non-bacterial) osteomyelitis in childhood
-
Epidemiology
-
very rare, non-infectious inflammation with multifocal bony lesions and periodic exacerbations of painful swellings
-
synonym: chronic recurrent multifocal osteomyelitis (CRMO)
-
pediatric equivalent of SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, osteitis)
-
prevalence: 1–2:1,000,000
-
age peak around 10 years [69]
-
-
Etiology
-
multifocal bony lesions
-
frequent association with other chronic inflammatory diseases, e. g., rheumatoid arthritis (RA), Crohn’s disease, sacroiliitis, psoriasis, or gangrenous pyoderma [70]
-
-
Clinical findings in cases of orbital involvement
-
cephalgia
-
hyperemic swelling
-
-
Diagnostics
-
CT scan: osteolytic findings
-
scintigraphy: focus with increased uptake
-
histology: lesions with osteoblastic/osteoclastic remodeling, granulomatous infection infiltrates with giant cells
-
biopsy of involved bone needed for confirmation of diagnosis and exclusion of differential diagnoses (e. g., Langerhans cell histiocytosis, osteoblastoma, osteosarcoma)
-
-
Therapy
-
first choice: NSAIDs (e. g., naproxen)
-
alternative: bisphosphonate or biologicals such as TNF antagonists (etanercept) or IL-1 inhibitors (anakinra)
-
-
Prognosis
-
unfavorable course of CRMO is possible (in contrast to earlier opinions) with persistent chronic pain and physical impairment in up to 50% of cases [71]
-
-
Important differential diagnosis: infectious osteomyelitis of the orbit
-
another rare disease in the area of the orbit
-
etiology: mostly after previous trauma, inflammation (paranasal sinuses, dentogenic, or secondary after bacteremia in the context of intravenous drug abuse or leukemia) [72]
-
Clinical: in acute stages, fever and swelling; in chronic courses, sequesters and fistulas
-
many other possible differential diagnoses, including bacterial cellulitis, atypical pathogens (tuberculosis), neoplasms as in Ewing sarcoma
-
#
#
5.1.2 Orbital fungal infections
Fungal infections as the origin of orbital inflammation are quite rare; however, this aspect should always be considered in immunocompromised patients. The most frequent infections are caused by Phytomycetes (Mucor spp.) and Ascomycetes (Aspergillus spp.).
5.1.2.1 Rhino-orbital mucormycosis
Mucormycosis is a rare infectious disease with high mortality. There often is a delay to diagnosis, and the disease is characterized by quick deterioration. Rapid surgical and medical treatment may be lifesaving [73].
-
Pathogenesis
-
ubiquitously occurring pathogen that leads to endothelial damage, ischemia, and the typical necrotic eschar (missing in early stages) because of angioinvasion
-
affecting the lung, skin, and gastrointestinal tract, but most frequently the paranasal sinuses with involvement of the orbit and finally also intracranial involvement via the orbital apex/lamina cribrosa
-
-
Etiology
-
almost always associated with another factor, e. g., uncontrolled diabetes mellitus (especially in cases of ketoacidosis), malignancies, burn injuries, hemochromatosis, post kidney or stem cell transplantation, intake of the chelating agent deferoxamine, AIDS
-
-
Clinical
-
general symptoms: fever, fatigue, pharyngitis
-
rhino-orbital symptoms: acute pansinusitis with purulent rhinorrhea, pain, and massive crust formation (black crusts at the nose and palate), subsequent apex-orbital syndrome with sudden loss of vision and ophthalmoplegia, stupor with intracranial extension with cavernous sinus thrombosis [58]
-
-
Diagnostics
-
suspected mucormycosis and rapid biopsy examination in cases of unclear facial complaints, paresthesia, swellings, and rapidly progressive sinusitis in patients with relevant comorbidities [74]
-
-
Therapy
-
pharmacotherapy of first choice: liposomal amphotericin B
-
other pharmacotherapy: isavuconazole and posaconazole, especially for salvage therapy [73] [75]
-
repeated and extensive debridement almost always required
-
exenteration necessary when the orbital apex is affected, but not mandatory in cases of affected anterior orbital parts [76]
-
-
Prognosis
-
formerly almost always lethal, but survival rate of ~70% with the introduction of amphotericin B [58]
-
in recent years, lethal courses observed in immune-healthy patients – especially after injuries or insect bites in warm climates – because of mucor infections caused by a new pathogen strain (Apophysomyces elegans) [58]
-
#
5.1.2.2 Aspergillosis of the orbit
Aspergillus spp. are ubiquitous and generally colonize the mucosa of the upper airways as harmless saprophytes. As opportunistic pathogens, they play a major role in immunocompromised patients (e. g., after kidney transplantation).

-
Classification: two types of courses are differentiated regarding infections of the head and neck:
-
non-invasive
-
because of a pre-existing lesion of the mucosa (mostly in the context of chronic sinusitis), colonization with formation of so-called fungus balls or chronic allergic rhinosinusitis
-
occurrence also in healthy patients
-
rarely affecting the orbit [77]
-
-
invasive
-
in cases of impaired immune competence (e. g., because of leukemia or AIDS), rapid distribution by vascular invasion with tissue necrosis, via ethmoid and sphenoid sinus into the orbital apex, the cavernous sinus, and in the intracerebral direction; development of thromboses and aneurysms with high mortality [78] [79] [80]
-
possible hematogenous dissemination from pulmonary infection foci or because of contaminated needles with heroin abuse [61]
-
-
-
Clinical
-
invasive type: similarly fulminant manifestation as for mucormycosis
-
non-invasive type: mostly slowly progressive indolent mass developing in the paranasal sinuses with extension in direction of the orbit, often pre-existing sinusitis
-
isolated involvement of the sphenoid sinus with optic neuritis without involvement of the orbit is possible [81] [82]
-
-
Diagnostics
-
rather low sensitivity and specificity of the single test procedures, warranting identification by a combination of imaging (CT scan, MRI), microbiological examinations (direct microscopy, culture, PCR, biomarker), and histopathology (branch-like septated hyphae, chronic-fibrous granulomatous inflammation) [83]
-
because of non-specific symptoms (minor sinusitis, retrobulbar, partly neuropathic pain, apical orbital inflammation difficult to identify by imaging), particular attention is needed in the context of diagnostic procedures [59]
-
-
Important differential diagnoses
-
bacterial cellulitis, cavernous sinus thrombosis, Tolosa-Hunt syndrome
-
tumors of the orbital apex or the sphenoid sinus
-
-
Therapy
-
antifungal therapy with liposomal amphotericin B until exclusion of mucormycosis, followed by a shift to the more effective voriconazole [84]
-
extensive debridement of the infected tissue, especially in the context of the invasive type
-
-
Prognosis
#
5.1.2.3 Other mycoses
Outside of Europe, other pathogens may be responsible for fungal infections of the orbit [58] [59], including:
-
North American blastomycosis
-
Fusariosis
-
African histoplasmosis
-
Sporotrichosis
-
Rhinosporidiosis
-
Coccidioidomycosis
-
Candidiasis
-
Bipolaris hawaiiensis
#
#
5.1.3 Viral infections
Orbital effects from viruses are rare; however, these microbes may cause diseases based on infection or secondary immune phenomena, including:
-
Varicella zoster virus (VZV): acute myositis, optic neuritis
-
Mumps virus: acute dacryoadenitis
-
Epstein-Barr virus (i. e., EBV): acute dacryoadenitis, some cases associated with T cell lymphomas of the orbit and the paranasal sinuses [61]
#
5.1.4 Parasitoses
Infestation of the orbit is also possible by larvae of different parasitic worms. In Germany, echinococcosis ranks among these rare diseases of the orbit.
5.1.4.1 Cystic echinococcosis
-
Epidemiology
-
occurring worldwide, especially when humans and animals live in close proximity
-
mainly patients between the 1st and 4th decades of life [59]
-
endemic distribution in Central and South America, Middle East, North Africa, China, and Russia
-
-
Pathogenesis
-
excretion with feces of eggs of adult tapeworms from the gut of carnivores (e. g., dogs)
-
intake of eggs by herbivores (e. g., sheep, cattle) as intermediate hosts or humans via the consumption of contaminated vegetables
-
distribution of the eggs in intermediate host via the blood circulation into the tissue and intake by the final host with food
-
growing in the final host until the final larval stage
-
effects in the lung and liver are most frequent, with orbital involvement in 1% of cases [88] [89]
-
-
Clinical
-
slowly progressive, indolent, intraconal mass
-
possible thinning of the orbital wall with intracranial extension
-
rupture of the cystic wall (spontaneous or in the context of surgical excision) may lead to fulminant inflammatory or anaphylactic reaction [61]
-
-
Diagnostics
-
Therapy
-
classic: surgical excision
-
alternative: different recommendations [91] [92], e. g.:
-
monotherapy with albendazole over 3 months; in cases of concomitant inflammation additionally systemic corticosteroids
-
combination of albendazole, praziquantel, and surgical excision
-
-
in cases of unclear diagnosis or compression-induced loss of vision, surgical excision required [61], fine needle aspiration may be applied for diagnostics and therapy (albendazole injection) [59]
-
#
5.1.4.2 Cysticercosis
-
Epidemiology
-
endemic occurrence in countries with poor sanitary conditions (Latin America, parts of Asia, and in South Sahara)
-
-
Pathogenesis
-
caused by the parasite Taenia solium (pork tapeworm)
-
intake of eggs via underdone pork and hematogenous distribution
-
formation of encapsulated larvae with preferential colonization in the eye and brain (neurocysticercosis, life-threatening) [61]
-
-
Clinical
-
Therapy
-
surgical excision and pharmacotherapy with albendazole or praziquantel
-
reduction of the concomitant inflammation by steroids [95]
-
#
5.1.4.3 Trichinosis
-
Pathogenesis
-
intake of encapsulated larvae by ingestion of underdone pork
-
in the gut, development of larvae into roundworms, development of new larvae, and hematogenous distribution
-
development of cysts in the striated muscles
-
-
Clinical
-
first manifestation often in the extraocular muscles
-
preseptal edema, chemosis, and painful myopathy
-
-
Therapy
#
5.1.4.4 Ophthalmomyiasis
-
Etiology
-
caused by botfly larvae in tropical regions
-
-
Clinical
-
the symptoms vary from mild conjunctivitis to destruction of the entire orbit
-
-
Therapy
-
removal of the larvae
-
disinfection up to exenteration in cases of severe disease
-
#
5.1.4.5 Other parasitoses and fly larvae with possible eye involvement [96]
-
Cestodes
-
Dirofilaria
-
Loa loa
-
Onchocerca volvulus
-
Oxyuris
-
Plasmodia
-
Porocephalus armillatus
-
Schistosoma
-
Thelazia
-
Wuchereria bancrofti
#
#
#
5.2 Non-infectious orbital inflammation
5.2.1 Introduction
In the proper sense of the word, orbital inflammation is not a diagnosis [97]. The inflammation corresponds to the adaptive response of the immune system to a cause, e. g., an infection or injury, and to autoimmune disease. In 20% of tumoral lesions of the orbit, an association with inflammation is found [98]. The detection of the underlying disease is often a major challenge. Common characteristics are signs of clinical inflammation as well as the histological manifestation of a polymorphic inflammatory cell infiltration [59]. With regard to occurrence and intensity, a significant variability is observed [99]. Endocrine orbitopathy as the most frequent non-infectious inflammatory change in the orbit will not be discussed because it falls outside the scope of this article covering rare diseases of the orbit.
#
5.2.2 Idiopathic orbit inflammation
Idiopathic orbit inflammation (IOE) was formerly known as ‘pseudotumor orbitae’. encompasses a heterogeneous group of diseases characterized by orbital inflammation without an identifiable local or systemic cause. It is a rare entity as well as an exclusion diagnosis [100], characterized by an increase in mass of orbital structures, which may be caused by non-specific inflammatory, sometimes fibrotic, and rarely even destructive lesions [61]. The pathophysiology is not fully clarified; an autoimmune genesis that might be triggered by infections has been proposed [99]. Several classifications have been suggested, but none are generally acknowledged because of the highly variable clinical and pathologic properties [101] [102] [103].
IOE may affect every orbital structure, and the first manifestation varies from acute onset to a slowly progressive course.
Depending on the location, the following differentiations can be made [61]:
-
Idiopathic dacryoadenitis: isolated and affecting the lacrimal gland (most frequent variant)
-
Idiopathic orbital myositis: limited to one or more extraocular muscles
-
Diffuse IOE affecting different orbital structures (rare)
-
Idiopathic perineuritis of the optic nerve (very rare): involvement of the optic nerve sheath
-
Anterior IOE: listed as an independent entity by some authors [103]
The list of differential diagnoses is long and encompasses almost all types of infectious and non-infectious orbital inflammation as well as a multitude of neoplastic and vascular lesions [99]. The diagnosis can be made only based on histology so that biopsies play a major role, especially in cases of advanced disease and recurrences, to exclude relevant differential diagnoses. Exceptions are idiopathic myositis with its characteristic clinical and radiological appearance as well as lesions in the area of the orbital apex where the risks of iatrogenic injuries are higher than the expected benefit. Good response to corticosteroids is observed in numerous different orbital lesions including endocrine orbitopathy and malignancies, so that this response should not be used for confirming the diagnosis.

-
Histological characteristics [100] [102]
-
classic orbital pseudotumor
-
broad inflammatory substrate of lymphocytic, granulocytic, and sometimes histiocytic infiltrates, organized into lymph follicles with germinal centers
-
increased connective tissue with edema and fibrosis
-
early visible destruction of the lacrimal gland acini or muscle fibers
-
delimitation to lymphoproliferative diseases is important
-
-
idiopathic sclerotic type
-
tissue sclerosis and hyalinization dominant
-
poor inflammatory infiltrate, similarity to retroperitoneal fibrosis
-
diagnostics based on IgG4-associated diseases recommended [104]
-
-
rarely granulomatous (histiocytic giant cell infiltration) and vasculitic (vasculitis of the minor vessels) inflammatory reactions
-
possible transition of classic orbital pseudotumors to the sclerotic subtype [100]
-
-
Clinical
-
highly variable, typically unilateral occurrence with disease history of several days to weeks, ranging from mild signs of inflammation to severe pain and increased orbital pressure with the risk of optic nerve compression; often recurrences in cases of classic pseudotumors as well as the sclerotic type [100]
-
depending on the location, painful (swelling-related) pseudoptosis, chemosis, exophthalmos, and (mostly pain-related) impaired ocular motility
-
sclerotic type: pain frequent, rather notable swelling of the eyelid, impaired ocular mobility, loss of vision ([Fig. 9a, b])
-
-
Imaging and serologic diagnostics
-
idiopathic dacryoadenitis/idiopathic myositis: increase in affected structures with unchanged differentiation
-
sclerotic type: homogeneously enhancing, poorly delimited infiltrates of different orbital structures (initially mostly lateral with involvement of the lacrimal gland), in the further course, possible extension in the direction of the cavernous sinus and pterygopalatine fossa; in addition, possible multifocal fibrosclerosis (in particular retroperitoneal) ([Fig. 9c, d])
-
broad laboratory and serological diagnostics for differential diagnosis and assessment of the inflammatory activity possibly useful (among others, kidney and thyroid values, ANCA, ACE, rheumatoid factors, IgG4 serum level)
-
-
Therapy
-
in cases of a high level of suffering, immediate therapy is required
-
different levels of anti-inflammatory therapy, from antibiotic therapy, NSAIDs, or steroids to application of immunosuppressants [100]
-
mostly good response to systemic high-dose therapy with corticosteroids, although response can be poor to corticosteroids and irradiation in cases of sclerotic type
-
with rapid diagnosis and onset of combination therapy consisting of corticosteroids and immunosuppressants (in cooperation with rheumatologists), delayed progression at least possible in cases of the sclerotic subtype [59]
-
in cases of refractory and chronic courses, systemic application of immunosuppressants (e. g., methotrexate and azathioprine), biologicals such as rituximab, and a combination with radiation of the orbit
-
surgery such as debulking in the lacrimal type, decompression of the orbit in cases of optic nerve compression, orbital exenteration as ultima ratio in persistent severe pain [99]
-
5.2.2.1 Tolosa-Hunt syndrome
-
Epidemiology
-
rare idiopathic granulomatous inflammation of the cavernous sinus and the orbital apex with compression of the neighboring cranial nerves (III–VI)
-
incidence of 1:1,000,000 per year [105]
-
-
Clinical
-
unilateral ophthalmoplegia and (partly migrating) pareses of the cranial nerves running along the cavernous sinus and the superior orbital fissure (the oculomotor nerve most frequently affected)
-
often associated with constant retrobulbar facial pain
-
-
Diagnostics
-
confirmation of the diagnosis by means of MRI or biopsy to exclude differential diagnosis (sarcoidosis, malignancies, cavernous sinus thrombosis, cavernous carotid fistula) [106]
-
-
Therapy
-
Prognosis
-
rapid pain relief from corticosteroids
-
Cave: even in cases of malignancies, high-dose corticosteroid application may lead to temporary improvement of the complaints; see case report in [Fig. 10a–f]
-
regression of neuropathies often only after several months, sometimes persisting
-
Case report 1

-
History
-
79-year-old male patient with right-sided periorbital headaches progressing for months: intermitting neuralgic character in the area of the ophthalmic nerve, touch-sensitive
-
trochlear and abducent paresis on the right side for weeks (diplopia)
-
-
Diagnostics
-
MRI of the orbit: contrast-enhancing structure in the extraconal space at the orbital roof (most likely thickened supraorbital nerve) reaching into the superior orbital fissure, focal contrast enhancement in the orbital apex and into the cavernous sinus ([Fig. 10a–c])
-
PET-CT: no glucose consumption typical for malignancy, in particular in the area of the orbit
-
lumbar puncture: no hint of a florid inflammatory reaction of the CNS, no local immunoglobulin synthesis, no oligoclonal bands, no hint on serology of acute infection with neurotropic pathogens
-
-
Further course
-
working diagnosis: suspected Tolosa-Hunt syndrome; differential diagnostics in direction of infectious genesis, systemic granulomatous inflammation, or neoplasm
-
interdisciplinary board: therapy attempt with systemic glucocorticoid application (also because of the significant periprocedural risk in case of biopsy), and analgesics
-
initial improvement (in particular of the bulbar motility) and regressive MRI findings; differential diagnosis of autoimmune genesis, malignancy could not be excluded with certainty
-
continuation of glucocorticoid therapy
-
because of pre-damaged lung (fibrosis) with immunosuppressive therapy, repeated pneumonia and/or influenza pneumonitis
-
after 3 months, indication for biopsy because of persistent complaints despite regressive MRI findings
-
histological diagnosis: poorly differentiated adenocarcinoma
-
further staging diagnostics without clear findings, thus most likely primary tumor location
-
-
Therapy
-
radiotherapy (with consideration of the comorbidities and patient’s wishes)
-
Even in cases of inflammatory processes of the orbit, malignant disease must be considered. Improvement with glucocorticoid therapy does not exclude malignancy.
#
5.2.2.2 IgG4-associated orbitopathy
-
Definition
-
subacute to chronic courses of immunological systemic disease with coarse lymphocytic infiltrates and fibrotic lesions [111]
-
-
Locations and clinical
-
most frequent manifestations are autoimmune pancreatitis, sclerotic cholangitis, nephritis and renal fibrosis, and involvement of the lacrimal glands, salivary glands, and lymph nodes [111]
-
manifestations in the area of the orbit are typical, each structure may be affected, but in particular:
-
lacrimal gland: IgG4-associated dacryoadenitis with mostly painless swelling, mostly bilateral manifestation and with sicca symptoms [112]
-
orbital nerves and muscles: mainly infraorbital nerve; mostly painless with diplopia
-
orbital fatty tissue: associated with increased lacrimal glands [112] [113]
-
rarely, destructive lesion with enophthalmos [113] [114], affecting ocular structures or the bony limits
-
in more than 70% of patients extraocular manifestations [111]
-
visual disorders as leading symptom [111]
-
-
-
Diagnostics
-
imaging
-
diffusely increased lacrimal gland, muscles (mainly lateral), infraorbital nerve, partly with swelling of the bone channel ([Fig. 11a, b])
-
often ipsilateral effect on the paranasal sinuses [112]
-
-
histology
-
lymphoplasmacellular infiltration with obliterating phlebitis and eosinophilic inflammation
-
percentage of IgG4-positive plasma cells>40% (triggering stimulus for differentiation of B lymphocytes in IgG4-producing plasma cells still unknown)
-
fibrosis in the further course
-
histologic diagnostic criteria: IgG4/IgG ratio>50% as well as>30% IgG4-positive cells per high-power field [115]
-
-
laboratory
-
increased IgG4 serum level, although non-specific (increased also in Churg-Strauss syndrome, sarcoidosis, allergic diseases) and not identifiable in particular in limited orbital involvement
-
otherwise, no signs of systemic inflammation [99]
-
-
-
Therapy
-
first choice: traditionally oral glucocorticoid application (30–40 mg/d) as induction therapy, low-dose maintenance therapy over years, regression of complaints often observed
-
recommendation from recent studies: application of rituximab not only in recurrences but also in first-line therapy, leading to significantly better outcome rates than other disease-modifying anti-rheumatic drugs (DMARDs), such as methotrexate, mycophenolate, and azathioprine [116]
-
surgical debulking and radiotherapy only rarely indicated [116] [117]
-
Case report 2

-
History
-
75-year-old patient, condition after functional endoscopic sinus surgery (FESS) in 2007
-
6 months prior, right eye swelling and diplopia for 6 months, re-FESS in a new location with a clinical finding of “orbital abscess”
-
3 months later, re-FESS with polypous pansinusitis with orbital involvement, also performed alio loco
-
at first in domo presentation, persistent diplopia, loss of vision, eyelid edema, hypoesthesia
-
-
Diagnostics
-
MRI: extraconal mass at the orbital floor not delimited from the inferior rectus muscle with contrast enhancement ([Fig. 11a, b])
-
after biopsy via a transconjunctival access, diagnosis of IgG4-associated orbital inflammation
-
-
Therapy (in cooperation with rheumatologists)
-
glucocorticoids and methotrexate, leading to full recovery
-
#
#
5.2.3 Vasculitis of the orbit
Orbital inflammation is often part of a systemic disease that cannot always be identified initially, even though it can have far-reaching consequences. Vasculitis plays a particular role. Vasculitic diseases encompass numerous pathologies; for the orbit, these are mainly orbital granulomatosis with polyangiitis (GPA, formerly called Wegener’s disease) as well as polyarteritis nodosa. These and other related diseases are listed below with their main characteristics.
5.2.3.1 Small and medium-sized vessel diseases
5.2.3.1.1 Orbital granulomatosis with polyangiitis (GPA)
-
Definition
-
necrotizing granulomatous inflammation of the respiratory tract and necrotic vasculitis of small and medium-sized vessels of the airways and kidneys
-
-
Epidemiology
-
Classification into two clinical types [119]
-
limited form
-
mostly affecting the upper airways
-
mostly no identification of ANCA
-
-
systemic form
-
-
Symptoms [111]
-
proptosis as a key characteristic (in combination with pulmonary and renal symptoms, groundbreaking for correct diagnosis)
-
painful ophthalmoplegia because of extension of the orbital mass into neighboring structures (e. g., cavernous sinus)
-
epiphora from obstruction of the efferent lacrimal ducts in cases of sinus involvement, diplopia caused by vasculitis of the extraocular muscle vessels, inflammation of the lacrimal gland, bulb perforation caused by necrotic scleritis, xanthelasma-like staining of the eyelids (“yellow lid sign”)
-
vision-threatening compression of the optic nerve in cases of infiltration of the neighboring structures
-
mostly subacute onset, mostly bilateral in the further course, episodes with significant progression of findings in the context of chronic disease
-
possibly dramatic deterioration during the disease course because of fibrosis and orbital atrophy with enophthalmos, motility disorders, and chronic pain [118] [121]
-
-
Diagnostic imaging (CT and MRI) [122]
-
extraconal infiltrates, mainly infero-medial in close proximity to the affected sinuses with bone destruction and septum perforation
-
possible isolated effect on the orbital apex (partly with intracranial extension) or isolated effect on the lacrimal gland (diffuse enlargement)
-
diffuse extension in intra- and extraconal direction (“wall-to-wall” tumor) as a pathognomonic sign of advanced stage
-
MRI suitable for identification of granulomatous inflammatory reaction
-
-
Pathology
-
classic histological triad of vasculitis, necrosis, and granulomatous inflammation in the orbit in 50% of patients
-
other characteristics: polymorphic inflammatory infiltrates, degeneratively changed granules/collagen as well as increased number of IgG4-positive plasma cells in many cases [123]
-
also in ANCA-negative GPA: additional immunohistochemical examination of IL-17 and IL-23 for differentiation of sarcoidosis [111]
-
-
Therapy
-
management of orbital GPA similar to that for systemic GPA
-
current standard: combination of corticosteroids with cyclophosphamide, methotrexate, or azathioprine; early therapy start may avoid fatal local or systemic courses [118]
-
in refractory cases: rituximab [124] (more favorable side-effect profile with comparable effect)
-
surgical debulking in cases of persistent proptosis, uncontrollable pain, or compressive neuropathy of the optic nerve [121]
-
#
5.2.3.1.2 Allergic granulomatosis (Churg-Strauss syndrome)

-
Definition
-
allergic systemic disease; initial manifestation with asthma and rhinosinusitis, and in the course, necrotic vasculitis of small and medium-sized vessels and eosinophilic granulomatous inflammation [118]
-
orbit rarely affected
-
-
Location
-
predominantly involving the lung, paranasal sinuses, and peripheral nerve system
-
-
Clinical
-
ischemic vasculitis or orbital inflammatory pseudotumor with dacryoadenitis, myositis, periscleritis, perineuritis
-
may be a first manifestation of the syndrome [125] ([Fig. 12a–c])
-
in the course, proptosis, diplopia, ophthalmoplegia, loss of vision from optic nerve compression
-
in the stage of vasculitis, life-threatening complications possible (peritonitis from gut perforation, eosinophilic myocarditis with myocardial infarction [127]
-
Diagnostics
-
biopsy of affected structures
-
main histological features: necrotizing vasculitis with extravascular Infiltrates ([Fig. 12d])
-
seropositive pANCA in 70% of patients [126]
-
imaging (CT, MRI): non-specific changes with increased retrobulbar space, swelling of lacrimal gland and extraocular muscles (EOM)
-
diagnostic criteria of American College of Rheumatology (1990; 4 of 6 must be applicable): asthma, paranasal sinus abnormalities, pulmonary infiltrates, eosinophilia greater than 10% of white blood cell differential, extravascular eosinophils, presence of a mononeuropathy or polyneuropathy [127]
-
-
-
-
Therapy
-
first-line therapy: systemic corticosteroids (oral, intravenously applied), additional methotrexate and cyclophosphamide as steroid-saving measure in cases of critical organ involvement
-
biologicals in case of recurrence or refractory disease: rituximab, infliximab, mepolizumab, or omalizumab [118] [128]; in general, permanent pharmacotherapy needed
-
-
Prognosis
-
before the era of biologicals: mortality in up to 50% of cases if two or more organs affected [129]
-
#
5.2.3.1.3 Polyarteritis nodosa
-
Definition
-
necrotic vasculitis of small and medium-sized arteries and sometimes of the accompanying veins
-
segmental effects characteristic (typically bifurcations)
-
typically thromboses and nodular aneurysms [59]
-
histological characteristics: fibrinoid necrosis of the media and simultaneous occurrence of different inflammatory stages
-
-
Epidemiology
-
mostly male patients in the 4th and 5th decades of life
-
association with hepatitis B
-
orbit involved in about 10% of patients [118], sometimes as the first manifestation of the disease
-
-
Location
-
predilection for kidney, heart, liver, gastrointestinal tract, peripheral nerves, and CNS, and also bones and joints
-
-
Clinical
-
typical ophthalmological lesions: retinal vasculitis, choroidal infarction with exudations, ischemic optic neuropathy
-
secondary phenomena of vasculitis: ischemia of the EOM with ophthalmoplegia, diffuse orbital inflammation with exophthalmos, impaired motility, chemosis
-
-
Diagnosis based on the combination of orbital inflammation with other disease symptoms, non-specific inflammation signs on labs, biopsy, and imaging (sometimes angiographic confirmation of aneurysms)
-
Therapy
-
systemic corticosteroids and cyclophosphamide
-
application of other DMARDs such as methotrexate, azathioprine, and rituximab also reported [118]
-
-
Prognosis
-
5-year survival rate of about 80% [118]
-
#
#
5.2.3.2 Vasculitis of the major vessels
5.2.3.2.1 Giant cell arteritis (temporal arteritis)
-
Definition
-
idiopathic vasculitis of the middle and major arteries of the head and neck
-
histologically segmental inflammation with giant cells, lymphocytes, plasma cells, and eosinophils [118]
-
-
Epidemiology
-
age peak in the 7th to 8th decades of life
-
mostly female patients [111]
-
orbital involvement rarely observed
-
-
Etiology
-
combination of genetic predisposition (European ancestry) and environmental factors (association with virus infection, e. g., VZV, CMV, parvovirus 19) [130]
-
-
Clinical
-
general symptoms
-
headaches, jaw claudication
-
B symptoms such as fever, fatigue, weight loss
-
polymyalgia rheumatica, scalp tenderness [111]
-
-
ophthalmological symptoms
-
loss of vision because of ischemia of the optic nerve (anterior ischemic opticus neuropathy [i. e., A-AION]: urgent treatment indicated)
-
further diplopia, choroidal and retinal ischemia signs [111]
-
-
orbital symptoms
-
ophthalmoplegia and diplopia (because of ischemia and inflammation) [131]
-
-
-
Diagnostics
-
diagnosis by means of combination of typical symptoms (headache, scalp tenderness), laboratory parameters (increased erythrocyte sedimentation rate (ESR), radiological findings (contrast enhancement of the orbit in MRI), and biopsy of the temporal artery (biopsy of the orbital lesion is rarely necessary) [118]
-
-
Therapy
-
first-line therapy: high-dose corticosteroids (Cave: undesired long-term effects)
-
current study results with tocilizumab (anti-IL-6) quite promising [132]
-
#
5.2.3.2.2 Takayasu arteritis
-
Definition
-
granulomatous panarteritis affecting the aorta and its main branches
-
-
Epidemiology
-
first description in middle-aged female Japanese patients
-
current investigations: all ethnic groups affected with increasing prevalence [133]
-
-
Clinical
-
vasculitic destruction of the carotid artery with collateral blood supply to the eye
-
possible loss of vision because of ischemic ocular complications
-
initially, amaurosis fugax or progressive permanent loss of vision possible [134]
-
-
Diagnostics
-
fluorescence angiography to confirm critical perfusion of the retina
-
in recent years, replaced by MR angiography and FDG-PET
-
-
Therapy
-
primary glucocorticoids
-
in steroid refractory cases: leflunomide and tocilizumab [135]
-
#
#
5.2.3.3 Vasculitis of vessels with variable size
5.2.3.3.1 Cogan’s syndrome
-
Definition
-
rare inflammatory systemic disease
-
main involvement of the visual and audiovestibular system as well as vessels of variable size [111]
-
-
Epidemiology
-
affects mostly young adults
-
age peak at 29 years
-
to date, about 250 cases published [136]
-
orbital involvement extremely rare
-
-
Etiology and pathogenesis
-
autoimmune genesis with origin in the inner ear
-
triggered by initial virus infection
-
identification of different autoantigens (CD 148, connexin 26) [137]
-
-
Classification
-
typical type
-
non-infectious interstitial keratitis with Menière-like vestibulo-cochlear disorder within 2 years after first manifestation [111]
-
-
atypical type
-
different ocular symptoms such as conjunctivitis, episcleritis, glaucoma, uveitis; labyrinthine affection about two years after first manifestation
-
partly severe hearing loss, bilateral deafness in 60% of the patients [138]
-
-
-
Clinical
-
resulting from necrotic vasculitis in about 70% of cases, systemic involvement with manifestation in the blood vessels (aorta, renal, and coronary vessels), neurological and gastrointestinal symptoms; severe, sometimes life-threatening course
-
in cases of orbital involvement (only case reports): hearing loss, vertigo, enophthalmos, and orbital inflammation (however, without biopsy confirmation of the genesis) [118]
-
-
Diagnostics
-
neurotological, ophthalmological, and angiological diagnostics to exclude differential diagnoses
-
-
Differential diagnoses
-
congenital syphilis, Susac’s syndrome, Vogt-Koyanagi-Harada syndrome
-
other vasculitis diseases, e. g., granulomatosis with polyangiitis [136]
-
-
Therapy
-
first-line therapy: systemic high-dose glucocorticosteroids, often leading to improvement in vestibulo-cochlear and ocular symptoms [139]
-
prophylaxis against recurrence: biologicals such as infliximab (application also as first-line pharmaceutic in combination with glucocorticoids at an early stage of the disease to avoid irreversible damage) [140]
-
#
5.2.3.3.2 Behcet’s disease
-
Definition
-
idiopathic systemic vasculitis of blood vessels of variable size with episodic course [141]
-
-
Epidemiology
-
highest prevalence in the Middle East and Asia
-
male:female ratio, 3.5:1
-
orbital involvement extremely rare
-
-
Etiology and pathogenesis
-
strong association with HLA-B51
-
non-specific increase in CRP and BSG [118]
-
-
Clinical
-
general symptoms
-
aphthous lesions of the oral mucosa and genitals
-
skin lesions and intraocular inflammation
-
further organ manifestations in the gastrointestinal tract, lung, muscles, joints, and CNS [111]
-
-
in cases of ocular involvement
-
mostly uveitis with hypopyon, retinal vasculitis
-
rarely, anterior ischemic optic neuropathy (i. e., AION)
-
Cave: ocular involvement requires particular attention because of the high morbidity
-
-
in cases of orbital involvement (single case reports)
-
affecting EOM and/or lacrimal gland (however, without biopsy confirmation) [142]
-
-
Diagnostics
-
Therapy
#
5.2.3.3.3 Kawasaki syndrome
-
Definition
-
systemic autoimmune vasculitis, preferentially affecting medium-sized vessels, in particular the coronary vessels
-
-
Epidemiology
-
children younger than 5 years
-
incidence: in Japan 240:100 000; in those of European descent, 9:100 000 [145]
-
-
Etiology and pathogenesis
-
histopathologically six typical lesions: endothelial degeneration, necrotic arteritis, granulomatous inflammation, media degeneration, scarring, development of aneurysms [118]
-
-
Clinical
-
cardinal symptoms: fever (>5 days), palmar and plantar erythema, polymorphic exanthema, bilateral conjunctival injection and chemosis, erythema of the oral mucosa, cervical lymphadenitis, aneurysms of the coronary vessels
-
ophthalmological symptoms
-
bilateral non-purulent conjunctivitis in 80% of cases with conjunctival injection and chemosis
-
anterior uveitis [145]
-
-
orbital inflammation (single case reports) confirmed by biopsy; otherwise, by diagnosis in the context of further symptoms of the disease [146] [147] [148]
-
-
Diagnostics
-
typical clinical constellation
-
important: cardiological diagnostics with heart echocardiogram, and coronary angiography if needed
-
lab: non-specific increase in ESR, CRP, alpha-1 antitrypsin, thrombocytes
-
-
Therapy
-
standard: high-dose intravenous application of immunoglobulins plus aspirin for symptom reduction and prevention of aneurysms of the coronary arteries [118]
-
therapy-refractory cases: infliximab [149]
-
application of corticosteroids is controversially discussed (administration only in refractory cases) [147]
-
#
5.2.3.3.4 Collagenosis-associated vasculitis: systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), dermatomyositis (DM)
-
Definition
-
Epidemiology
-
Etiology and pathogenesis
-
immunocomplex-mediated pathogenesis
-
-
Clinical
-
frequently: cutaneous lesions, involvement of the mesenterial, coronary, and pulmonary arteries, the arteries of the lower extremities, and the CNS
-
ocular involvement: mostly retinopathy, keratoconjunctivitis sicca, uveitis
-
single publications about symptoms of orbital inflammation (infarction, myositis, exophthalmos)
-
orbital inflammation symptoms together with according general symptoms and typical serological findings of SLE and RA (increased ANA and ds-DNA-AB) [151] [152] [153]
-
very rare orbital involvement in DM: case report of delayed bilateral orbital involvement with exophthalmos by increased EOM, associated with muscle weakness, and typical EMG changes (without confirmation by biopsy) [154]
-
-
Therapy
-
SLE and RA: glucocorticoids intravenous or oral; alternatively, azathioprine and biologicals (rituximab, infliximab, belimumab)
-
DM: glucocorticoids intravenous; also immunoglobulins as first-line therapy as well as methotrexate and azathioprine have been described [118]
-
#
#
#
5.2.4 Granulomatous inflammation of the orbit
5.2.4.1 Sarcoidosis
Sarcoidosis is a chronic inflammatory systemic disease. The term ‘sarcoid’ was introduced by Caesar W. Boeck because these lesions are histologically similar to sarcomas without being malignant. Effects in the lung with bihilary lymphadenopathy as well as skin and eyelid lesions are typical findings [155] [156] [157].
-
Etiology and pathogenesis
-
increased inflammatory reaction to some pathogens (not precisely identified) with increased cellular immune response and formation of non-caseating granulomas
-
genetic component (higher familial incidence)
-
-
Epidemiology
-
Location
-
ocular involvement (including the most frequent extrapulmonary symptoms): conjunctivitis, uveitis, chorioretinal lesions, vitreous body changes
-
orbital involvement possible as first manifestation: mostly lacrimal gland (often bilateral involvement); furthermore, orbital fatty tissue, optic nerve sheath (as part of neurosarcoidosis), efferent lacrimal ducts, or eye muscles [155] [156] [159] [160] [161]
-
-
Clinical
-
Diagnostics
-
biopsy essential for confirmation of the diagnosis; because of the histological similarity with other lesions such as idiopathic orbital inflammation or lymphoproliferative diseases, diagnosis can be difficult
-
serology: angiotensin converting enzyme and liver values; possibly inconspicuous serological findings
-
imaging
-
MRI: if affected, the lacrimal gland is diffusely increased with homogeneous enhancement, with further manifestations as a poorly delimited mass, swelling of the optic nerve sheath, or diffusely increased muscles
-
thoracic X-ray for pulmonary assessment
-
-
-
Therapy
-
intravenous corticosteroids to improve the functional prognosis (essential)
-
interdisciplinary care with rheumatologists is important
-
surgical excision or debulking if needed, but only in combination with previous systemic therapy [159] [161]
-
in refractory cases or recurrence: immunosuppressive combination therapy, e. g., with methotrexate or azathioprine, or with biologicals such as infliximab or adalimumab [156] [161]
-
#
5.2.4.2 Melkersson-Rosenthal syndrome
-
Definition
-
neuromucocutaneous granulomatous dermatosis
-
-
Epidemiology
-
incidence of 0.08% [162]
-
first manifestation mostly in young adults
-
-
Etiology
-
autosomal dominant inheritance with variable expression [163]
-
-
Clinical
-
classic symptom triad: lingua plicata, cheilitis granulomatosa, peripheral facial nerve palsy; mono- or oligosymptomatic appearance quite frequent
-
often diffuse edematous facial swellings, rarely isolated in the area of the eyelids (thus a difficult diagnosis, especially when other characteristics are missing; imaging and biopsy may facilitate the delineation)
-
rare involvement of other cranial nerves [162] [163] [164] [165] (e. g., diplopia because of oculomotor paresis [162] [166])
-
-
Therapy
-
Prognosis
-
variable chronic course with spontaneous remissions and recurrences
-
#
5.2.4.3 Foreign body granulomas and idiopathic lipogranulomas

-
Definition
-
granulomatous inflammatory reaction caused by foreign bodies
-
-
Classification according to the type of foreign body
-
ruptured dermoid cyst ([Fig. 13a–d])
-
most frequent intrinsic cause of foreign body granulomas (partly with inflammatory reaction)
-
mostly inhomogeneous lesion with irregular borders on CT scans
-
therapy: excision (possibly complete removal without damaging neighboring structures; if small fibrotic parts remain, treatment with intralesional corticosteroids)
-
-
herbal foreign bodies
-
further source of granulomatous inflammatory reaction, exact history-taking is important
-
slowly progressive course and fistulation possible
-
-
paraffin-induced sclerotic lipogranuloma
-
mostly as consequence of application of moisturizing nasal ointments after endoscopic sinus surgery (with intraoperative damage of the lamina papyracea) [168] [169]
-
accidental leakage of intraocular silicone oil in the context of ophthalmological interventions
-
after autologous fat injection in plastic surgery
-
-
idiopathic lipogranuloma
-
in cases of missing trigger
-
only single case reports in the literature, unclear etiology
-
histologically orbital fatty tissue necrosis [170]
-
-
-
Clinical
-
eyelid swelling
-
subcutaneously palpable nodes
-
in cases of tumoral lesions: exophthalmos, impaired oculomotor function, and bulb deviation are possible
-
-
Diagnostics
-
Therapy
-
complete surgical removal with subsequent systemic glucocorticoid application is most effective [170]
-
optionally: triamcinolone injection or debulking
-
-
Prognosis
#
#
5.2.5 Sjögren’s syndrome (SS)
-
Definition
-
chronic autoimmune disease characterized by inflammation of the lacrimal and salivary glands as well as the epithelia
-
keratoconjunctivitis sicca as one of the leading diagnostic criteria (inflammation of the accessory conjunctival lacrimal glands) [61]
-
-
Epidemiology
-
age peak in the 4th to 5th decades
-
affects mainly women
-
-
Classification
-
primary SS: independent disease with ocular and oral symptoms (Sicca syndrome)
-
secondary SS (more frequently observed): in the context of other autoimmune diseases (RA, SLE, systemic sclerosis)
-
-
Etiology and pathogenesis
-
environmental and hormonal factors as well as genetic predisposition
-
lymphocytic infiltration and sclerosis of the lacrimal and salivary glands by T cells and autoantibodies; B cells presumably also pathogenetically significant [175]
-
-
Clinical
-
slowly progressing, painless swelling of the lacrimal gland (and sometimes of the salivary glands)
-
in the further course, glandular atrophy, often bilateral, xerostomia, xerophthalmia (rather in the atrophic stage)
-
other ocular and orbital symptoms: uveitis, episcleritis/scleritis, optic neuropathy [155] [176], myositis of the EOM [177]
-
extraglandular symptoms: fatigue, pain, polyneuropathy, vasculitic skin lesions, pulmonary changes, musculoskeletal, renal, and gastrointestinal manifestations [175]
-
-
Diagnostics
-
CT/MRI: orbitally enlarged lacrimal glands with enhancement, difficult to delimit to lymphoproliferative diseases [178]
-
lab: increased specific antinuclear antibodies (SS-A and SS-B, the latter highly specific for SS)
-
biopsy: confirmation of the diagnosis and exclusion of lymphoma (SS patients have a 20-fold increased risk for lymphoma, in particular in the head and neck [175] [179] [180])
-
-
Therapy
-
interdisciplinary therapy is necessary because SS is relatively resistant against pharmacotherapy with corticosteroids and other anti-rheumatics
-
symptomatic therapy of sicca symptoms by ophthalmologists and systemic rheumatological medication consisting of steroids and immunosuppressants, application of biologicals, e. g., rituximab, mostly recommended in SS with extraglandular manifestation [181]
-
#
5.2.6 Kimura syndrome
-
Definition
-
benign chronic inflammatory disease of the subcutaneous tissue, of unknown genesis, also called angiolymphoid hyperplasia with eosinophilia
-
first description by Kimura in 1948 [182]
-
-
Epidemiology
-
mainly observed in males of Asian origin between the 20th and 30th years of life
-
orbit rarely affected
-
-
Histopathology
-
Clinical
-
Diagnostics
-
typical findings: increased serum IgE and blood eosinophilia
-
-
Therapy
#
5.2.7 Histiocytic diseases
Orbital xanthogranulomatous diseases are characterized by proliferation of histiocytes and summarized within the term ‘non-Langerhans cell histiocytosis’. In contrast, Langerhans cell histiocytosis has its origin in dendritic cells, associated with the Rosai-Dorfman syndrome and monocytes/macrophages. These diseases are described below.
5.2.7.1 Non-Langerhans cell histiocytoses
5.2.7.1 Juvenile xanthogranuloma
-
Definition
-
benign proliferative cutaneous disease, mainly affecting the eyes and skin
-
primary effects on the orbit with proptosis quite rare
-
-
Numerous differential diagnoses in childhood
-
Diagnostics: biopsy confirmation of diagnosis required
-
Therapy and prognosis
-
mostly spontaneous remission
-
in symptomatic patients with non-resectable lesions, corticosteroids and chemotherapeutics are applied
-
#
5.2.7.1.2 Adult-onset xanthogranuloma (AOX) and adult-onset asthma with periocular xanthogranuloma (AAPOX)

-
Etiology and pathogenesis
-
unknown, probably autoimmune genesis with deposits of immune complexes and subsequent foreign body reactions in the tissue
-
unknown cause of the affinity for orbital adnexa structures [188]
-
-
Clinical
-
yellowish infiltrates in the area of the conjunctiva and eyelids (xanthelasmas)
-
bilateral occurrence possible
-
-
Diagnostics
-
confirmation of the diagnosis by biopsy: histologically lipid-charged macrophages (xanthoma cells) in the dermis and deeper tissue layers, along with foreign body giant cells and Touton giant cells
-
association with hematological diseases (lymphoproliferative lesions, hepatosplenomegaly) possible, requiring thorough physical evaluation
-
determination of the orbital extension by CT to assess infiltration of the orbital fat, EOM, and lacrimal gland
-
in cases of association with asthma, the term ‘adult-onset asthma with periocular xanthogranuloma (AAPOX)’ is used [189]
-
-
Therapy
-
surgical debulking and plastic reconstruction
-
irradiation
-
systemic corticosteroids and other immunosuppressants ([Fig. 14a, b])
-
intralesional triamcinolone highly effective [190]
-
-
Prognosis
-
progressive disease
-
vision-threatening course without therapy
-
#
5.2.7.1.3 Necrobiotic xanthogranuloma
-
Epidemiology
-
rare disease occurring in patients between the 5th and 6th decades of life
-
-
Pathogenesis
-
foreign body giant cell reactions to deposits of lipid serum immunoglobulin complexes in the skin [188]
-
necrobiosis as histopathological key characteristic: eosinophilic degeneration of collagen in a granulomatous inflammation reaction with foamy histiocytes, foreign body giant cells, Touton giant cells, and lymphocytes
-
-
Location
-
development of infiltrates at the eyelids and ocular adnexa structures as well as the extremities
-
effects on inner organs and hematological neoplasms also possible [61]
-
-
Clinical
-
reddish-yellow lesions or flesh-colored nodules with extension into the anterior orbit with subsequent proptosis, ptosis as well as scar-related lagophthalmos
-
-
Therapy
-
many different therapeutic alternatives in use, including surgery, radiation, plasmapheresis, intralesional and systemic steroid application as well as cytostatic agents
-
case reports describing successful application of thalidomide in combination with dexamethasone [191]
-
overall, pharmacotherapy superior to radiation and surgical debulking
-
-
Prognosis
-
progression after biopsy or debulking possible
-
lifelong follow-up because of increased risk of hematological malignancy
-
#
5.2.7.1.4 Erdheim-Chester disease
-
Definition
-
rare type of non-Langerhans cell histiocytosis with progressive course, characterized by bone pain, retroperitoneal fibrosis, and infiltrations in the face
-
pathological characteristics: wide-spread infiltration of foamy (lipid charged) non-Langerhans cell histiocytes
-
-
Epidemiology
-
first manifestation mostly between the 5th and 7th decades of life
-
predominantly in males
-
more than 600 cases described since 1930 [192]
-
-
Etiology
-
not completely understood
-
association with different immunological diseases as hint for abnormal interaction between T cells and macrophages (uncontrolled activation of the macrophages by functional disorders of natural killer (NK)-T cells or cytotoxic T cells)
-
-
Clinical
-
characteristic: bone pain, especially in the distal lower extremities because of osteosclerosis of the long bones
-
often bilateral orbit effects: xanthogranulomas with partly deep retrobulbar infiltration, thus ophthalmoplegia and compressive optic neuropathy also possible in addition to exophthalmos
-
general symptoms such as fever and weight loss
-
severe disorders possible, including diabetes insipidus, cerebellar and pyramidal tract signs, cranial nerve paresis, adrenal insufficiency, pulmonary fibrosis, and cardiac decompensation [192] [193] [194]
-
variable course that can include fulminant organ failure
-
-
Therapy
-
Prognosis
-
poorest among all non-Langerhans cell histiocytosis with mortality rates of>60% within 3 years [196]
-
#
5.2.7.1.5 Disseminated xanthogranuloma
-
Definition
-
very rare, non-familial, histiocytic, infiltrative disease primarily affecting skin, mucosa, and hypothalamus
-
-
Epidemiology
-
about 100 cases reported
-
disease onset mostly in young adults [197]
-
male:female ratio, 2:1
-
-
Etiology and pathogenesis
-
causes or risk factors not known
-
characteristic: accumulation of histiocytes at the entire integument and CNS with accumulation of cholesterol and other lipids, leading to a histologically foamy appearance (typical for xanthomatous lesions)
-
-
Clinical
-
reddish cutaneous nodes, mainly at the surface of the flexors
-
cornea, conjunctiva, and eyelids affected (20%)
-
dysphagia or dysphony because of involvement of the upper airways (40%)
-
diabetes insipidus from meningeal involvement (40%)
-
-
Diagnostics
-
pathognomonic: triad of typical skin alterations, dysphagia, and diabetes insipidus [198]
-
bone marrow biopsy and serum electrophoresis required to exclude myeloid melanoma (observed in more than 50% of the patients)
-
histologically, not differentiable from Erdheim-Chester disease
-
-
Therapy
-
currently, causal therapy not possible
-
therapy attempts with numerous different medications, e. g., corticosteroids, lipid reducers, and several immunosuppressants; cladribine (purine-nucleoside analogue) currently most promising [199]
-
-
Prognosis
-
possible local control by surgical excision or CO2 laser ablation
-
#
#
5.2.7.2 Langerhans cell histiocytosis
-
Definition
-
Epidemiology
-
rare, predominantly occurring in children and adolescents
-
prevalence of 4–5:1,000,000
-
involvement of the orbit in 25% of cases
-
-
Etiology and pathogenesis
-
unclear, presumably clonal proliferation of phenotypically abnormal Langerhans cells
-
expression of atypical surface proteins and deposits in the tissue where Langerhans cells otherwise are not present (e. g., bones)
-
-
Types
-
unifocal manifestation: eosinophilic granuloma (most frequent type)
-
multifocal manifestation: Hand-Schüller-Christian disease with the triad of exophthalmos, diabetes insipidus, and osteolysis, very rarely Letterer-Siwe syndrome with progressive involvement of bone marrow and other organs [202]
-
-
Clinical
-
typical orbital findings: proptosis, ptosis, and papillary edema, bone destruction or the temporal orbital part
-
additional inflammatory reaction
-
-
Diagnostics
-
CT scan for detection of bone lesions
-
MRI to assess the cranial extension [203]
-
diagnosis based on typical clinical lesion, immunohistochemical identification of the surface markers of CD1a and CD207 as well as electron microscopic evidence of typical Birbeck granulae
-
-
Differential diagnoses in pediatric patients
-
acute infections (eyelid edema), inflammatory pseudotumor, dermoid cyst, rhabdomyosarcoma, Ewing sarcoma, osteosarcoma, or neuroblastoma, Erdheim-Chester disease
-
-
Prognosis
-
better with increasing age at first diagnosis
-
rather poor in cases of multifocal involvement
-
5-year survival rate maximum of 50% in cases of Letterer-Siwe syndrome despite modern chemotherapeutics
-
-
Therapy
-
local excision as therapy of choice in cases of isolated orbital involvement
-
intralesional injection of corticosteroids (prompt resolution described)
-
low-dose radiation in cases of lesions that cannot be approached surgically
-
systemic therapy for incomplete resection or with recurrence
-
prednisone for disseminated disease
-
chemotherapeutics in cases of poor response
-
-
hormone substitution in cases of hypopituitarism [61]
-
#
5.2.7.3 Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorman syndrome)
-
Definition
-
rare disease with massive infiltration of S-100 positive histiocytes in the tissue and cervical lymph nodes [204]
-
-
Etiology and pathogenesis
-
possibly viral genesis and genetic component
-
histopathologically characteristic: emperipolesis with phagocytosis of lymphocytes, erythrocytes, and neutrophilic granulocytes by large S-100 positive histiocytes
-
-
Epidemiology
-
Clinical
-
first manifestation mostly as indolent cervical lymph node swelling with accompanying fever, weight loss, and night sweats
-
extranodal manifestations in>40% of patients: eye, salivary glands, nose, bones, skin, or CNS [204] [207], necessitating systemic examination
-
in cases of massive lymph node swelling, possible life-threatening compression of the upper airways [208]
-
orbital manifestation: proptosis, swelling of eyelids or lacrimal glands
-
intra- or extranodal involvement: possible compressive neuropathy of the optic nerve and uveitis [208]
-
-
Therapy
-
generally self-limiting, indication for treatment mainly in cases of airway obstruction resulting from sudden size progression of massive cervical lymph node swellings
-
first-line pharmacotherapy: systemic high-dose therapy with corticosteroids
-
surgical intervention (excision or debulking) according to the severity of the orbital manifestation, further functional and esthetic corrections possible
-
in cases of idiopathic eyelid edemas, systemic corticosteroids or other immunosuppressants
-
in cases of life-threatening obstruction of the airways: surgical therapy or emergency radiation inevitable
-
chemotherapy if needed in cases of severe orbital involvement with optic neuropathy [204] [209]
-
#
#
5.2.8 Castleman’s disease
-
Definition
-
rare lymphoproliferative disease
-
-
Classification in two clinical categories
-
monocentric type
-
benign localized hyperplasia of lymphatic tissue
-
curative treatment by excision of the affected lymph nodes
-
-
multicentric type
-
-
Classification into three histological categories
-
hyaline vascular type (80–90%)
-
plasma cell type (10–20%)
-
rare mixed type
-
-
Location
-
extranodal involvement of the orbit quite rare, with few cases reported
-
monocentric variant with hyaline vascular type in more than 90% of the orbital cases
-
-
Clinical
-
mostly indolent swelling (proptosis)
-
in some cases, B symptoms
-
-
Diagnostics
-
Therapy
#
#
5.3 Conclusion
The large spectrum of possible diagnoses and the frequently overlapping clinical and radiological presentations of orbital inflammation emphasize the significance of biopsies for identifying the cause. In addition to the many infectious diseases that can entail possibly severe complications, autoimmune diseases often present initially in the orbit and must be assessed accurately with regard to serology and clinical manifestation. Among non-infectious inflammatory lesions, idiopathic orbital inflammation is most frequently observed. IgG4-associated diseases have gained in importance in recent years as a cause of orbital inflammation. Because of their similarity to lymphoproliferative diseases, non-infectious diseases of the orbit require thorough assessment. Histiocytic diseases remain important rare conditions that cause inflammation and fibrosis of the orbit and can lead to severe organ failure.
#
#
6. Degenerative Diseases: Orbital Amyloidosis
6.1 Orbital amyloidosis
The term ‘amyloidosis’ describes a heterogenic group of diseases characterized by extracellular accumulation of insoluble proteins in the β leaflet structure. Organ function may be disturbed by compression or direct cytotoxicity. Amyloidosis may present as a primary condition or secondarily as sequela of another disease. A genetic predisposition has been described [215] for this rare systemic or localized disease.


-
Location
-
deposits of amyloid possible in all structures of the orbit and the ocular tissue [216] [217] [218]
-
cases described with involvement of the lacrimal gland, eyelids, conjunctiva, and eye muscles including the palpebral levator muscle ([Fig. 15a, b])
-
-
Clinical
-
ptosis, exophthalmos, motility impairment with diplopia, bulbus deviation, ectropium or entropium, palpable tumor [218]
-
-
Differential diagnoses
-
chronic inflammatory gut diseases, tuberculosis, multiple myeloma
-
examinations required of heart, kidneys, gastrointestinal tract, and CNS [215]
-
-
Diagnostics
-
difficult diagnosis because of a lack of typical symptoms
-
MRI: inhomogeneous and contrast-enhancing deposits
-
CT scan: calcifications
-
histopathology: amyloid-typical apple-green staining as well as polarization with microscopically double-refractive properties
-
in cases of localized involvement, exclusion of a systemic variant required
-
-
Therapy
-
depending on the type, ranging from systemic chemotherapy with cortisone application to transplantation of the affected organs
-
in cases of localized orbital amyloidosis: surgical removal as therapy of choice if needed correcting interventions, e. g., for eyelid malpositions [215] [216] [218] [219]
-
radiotherapy as prophylaxis against recurrence recommended by some authors [216] [220]
-
-
Prognosis
-
highly variable course in cases of orbital involvement, from complication-free localized findings to severe organ damage with loss of vision [215]
-
#
6.2 Storage diseases [221] [222] [223] [224] [225]
Patients with storage diseases like Pompe’s disease or Gaucher’s disease have a high prevalence of clinically significant ophthalmological symptoms such as ptosis, bulbar motility disorders, strabismus, myopia, and astigmatism, in particular in classic infantile manifestations. These symptoms are mainly the result of the accumulation of glycogen (Pompe’s disease) or glucocerebroside (Gaucher’s disease), especially in the EOM, which may affect other ocular adnexa structures.
#
6.3 Mitochondrial diseases
6.3.1 Chronic progressive external ophthalmoplegia (CPEO)
-
Definition
-
very rare systemic muscle disease with slowly progressive paresis of all external eye muscles
-
-
Epidemiology
-
in addition to Leber’s hereditary optic neuropathy, the most frequently observed mitochondrial disease of adults with eye involvement
-
prevalence: about 12:100 000 [226]
-
onset mostly between the 20th and 50th years of life
-
-
Clinical
-
ptosis often the first symptom [227]
-
-
Differential diagnoses
-
other mitochondrial diseases, e. g., Kearns-Sayre syndrome (also ptosis and external ophthalmoplegia, onset mostly before the age of 20 years, additionally cardiac conduction disorders, cerebellar ataxia, and/or increased liquor albumin)
-
MELAS syndrome, (ocular) myasthenia gravis, oculopharyngeal muscle dystrophy
-
-
Diagnoses
-
MRI: atrophy of the extraocular muscles [228]
-
skeletal muscle biopsy: evidence of numerous “ragged red fibers” and COX-negative fibers
-
#
6.3.2 Leigh syndrome
-
Definition
-
neurodegenerative disease (mitochondriopathy) in children
-
-
Epidemiology
-
Etiology
-
genetically very heterogenic, up to now identification of mutations in 75 genes [231]
-
-
Clinical
-
highly variable symptoms
-
in the context of infection, development of neurological symptoms such as ataxia and dystonia and also ophthalmological symptoms, mainly strabismus, melanotic retinopathy, optic nerve atrophy, ptosis, and nystagmus [232]
-
later symptoms in other organs such as heart, liver, kidneys, or gastrointestinal tract
-
-
Prognosis
-
progress in phases, mostly with death in the 3rd year of life
-
#
#
#
7. Tumors of the Orbit [2] [201] [233]
7.1 General remarks
With an incidence of 6–10 per 1 million people, orbital neoplasms are rare, but they encompass a multitude of different entities that can lead to difficulties in the context of diagnostics. About 60% in adults are benign and 40% are malignant. “True” orbital neoplasms have to be differentiated from inflammatory orbital lesions without identifiable origin (idiopathic orbital inflammation, formerly called pseudotumors). They represent about 5–7% of all orbital masses and respond well to steroids and immunosuppressants. In the context of history-taking and diagnostics, the exact location, invasiveness, and status should be clarified. Diagnostics and therapy are often performed in an interdisciplinary context.
7.1.1 Symptoms
Neoplasm of the orbit may be classified as described below.
Because of the anatomical circumstances, typically the following symptoms are observed (with decreasing incidence) [201]:
-
Exophthalmos/bulbus protrusion (key symptom)
-
Bulbus dislocation
-
Motility disorders, diplopia
-
Pain
-
Loss of vision
-
Impaired visual field
Intraconal lesions tend to lead to axial exophthalmos, whereas extraconal masses tend to lead to bulbus dislocation to the contralateral side of the tumor (e. g., a tumor of the lacrimal gland leads to dislocation into the inferior nasal direction).
#
7.1.2 Classification and overview [2] [201]
Neoplasms of the orbit may be classified as follows:
-
Neoplasms of epithelial origin
-
Neoplasms of non-epithelial origin
-
Neoplasms of lymphatic tissue
In each category, benign and malignant entities are found. The only epithelial structure in the orbit is the lacrimal gland, and primary epithelial neoplasms thus occur there. Vascular lesions are sometimes not considered to be neoplasms but instead are viewed as malformations and have a special status. However, they may behave clinically like “true” neoplasms.
#
7.1.3 General therapeutic principles of benign neoplasms of the orbit [2] [201]
In general, the following therapy options are possible:
-
“Watch-and-wait” in cases of asymptomatic benign neoplasms without diagnostic uncertainty (e. g., incidentally discovered hemangioma)
-
Surgical resection as most frequently performed therapy in cases of symptomatic benign lesion
-
Other treatment strategies such as radiotherapy or chemotherapy in the context of specific entities
In the context of symptomatic benign processes, a thorough weighing of the expected therapeutic benefit against the risk of functional damage is necessary. Resection of an optic sheath meningioma, for example, is associated with a high risk of optic nerve atrophy.
#
#
7.2 Particularities
7.2.1 Neoplasms of the lacrimal gland
-
Epidemiology [233] [234] [235] [236]
-
incidence: 0.6–1:1 000 000 people per year; about 80 newly diagnosed diseases per year in Germany
-
tumors of the lacrimal gland encompass about 25% of orbital neoplasms
-
the distribution of tumors of different tissue types in the lacrimal gland is as follows [237]:
-
epithelial origin:>50–65%
-
lymphatic origin (special type of mesenchymal neoplasm): about 30–35%
-
mesenchymal origin or metastases: 10–15%
-
-
ratio of benign to malignant epithelial neoplasms is almost 1:1
-
-
Differential diagnoses [233]
-
inflammatory diseases
-
acute dacryocystitis
-
chronic dacryocystitis
-
idiopathic orbital inflammation (formerly called pseudotumor orbitae, dacryoadenitic type, representing about 20–40% of all cases)
-
-
structural changes (e. g., lacrimal gland cyst)
-
secondary tumors (not originating from glandular tissue, e. g., metastases, schwannomas)
-
-
General therapeutic strategies [235] [236]
-
for suspected epithelial tumor of the lacrimal gland, incision biopsy is contraindicated because of association with significantly worse prognosis of benign as well as malignant tumors
-
e. g., pleomorphic adenoma: 5-year recurrence rate of 32% after biopsy vs. 3% without biopsy prior to tumor resection
-
in addition, possible malignant degeneration of locally recurrent pleomorphic adenomas
-
lacrimal gland malignancy: 5-year survival rate of 29% after biopsy vs. 70% in cases of initially complete resection
-
-
precise preoperative diagnostic assessment desirable, based in part on symptom duration, pain, and radiological characteristics
-
the most effective and safe therapy in cases of epithelial tumors: complete resection during the first surgical intervention
-
complete resection indicated for encapsulated and well-circumscribed processes, without prior incision biopsy
-
surgical access via lateral orbitotomy and rarely in combination with coronal incision
-
7.2.1.1 Benign neoplasms of the lacrimal gland
[Table 3] summarizes the most important benign neoplasms of the lacrimal gland. Because of their specific characteristics, lymphoid neoplasms are also dealt with in the section on malignant diseases even if they are, strictly speaking, neoplasms of mesenchymal origin.
|
Tissue type |
Neoplasm |
|---|---|
|
Epithelial |
Pleomorphic adenoma |
|
Mesenchymal |
Solitary fibrous tumor |
|
Lymphoid |
Reactive lymphoid hyperplasia |
7.2.1.1.1 Benign epithelial neoplasms of the lacrimal gland
7.2.1.1.1.1 Pleomorphic adenoma [233] [234] [236] [238] [239] [240] [241] [242] [243] [244]
-
Epidemiology
-
most frequent epithelial tumor of the lacrimal gland (<50%)
-
about 20% of all lacrimal gland tumors [237]
-
manifestation in the 4th to 5th decades of life
-
male : female ratio, 1:1
-
-
Typical clinical findings
-
very slow growth
-
bulbus dislocation in inferior nasal direction (95%), impaired motility, sometimes with diplopia (40%), epiphora
-
-
Diagnostics
-
CT scan as diagnostic measure of choice
-
MRI provides only minor additional information (hyperintense signal in T2 weighting)
-
extraconal round-oval, sharply delimited lesion in the lateral superior quadrant of the orbit, inhomogeneous structure, displacing growth, dilation of the bony lacrimal gland fossa (81% in CT scan), calcifications as possible hint of malignant transformation
-
ultrasound: well-delimited, hypoechoic mass with dorsal sound amplification, low internal echo; on color duplex sonography, vascularization and perfusion identified only in the area of the capsule
-
-
Other diagnostic criterion
-
Biopsy
-
risk of incision biopsy doubted by some authors; however, most authors confirm this risk
-
benefit and risk of fine needle aspiration cytology are controversially discussed
-
-
Therapy
-
complete tumor extirpation without opening the tumor capsule with a margin of glandular tissue (access: lateral orbitotomy, palpebral glandular parts should be preserved)
-
after previous biopsy: enbloc resection of the scar with the biopsy pathway as well as the lacrimal gland
-
-
Prognosis
-
in cases of incomplete resection and incision biopsies or untreated status, risk of recurrence (32% within 5 years after incision biopsy vs. 3% after complete capsule-preserving resection) or malignant transformation (20% within 30 years)
-
|
Characteristic |
Score -1 |
Score+1 |
|---|---|---|
|
Duration of symptoms |
<10 months |
>10 months |
|
Persistent pain |
+ |
– |
|
Sensitivity disorder |
+ |
– |
|
Well-delimited, round-oval (CT scan) |
– |
+ |
|
Growth along orbital structures (CT scan) |
+ |
– |
|
Calcifications (CT scan) |
+ |
– |
|
Bone destructions (CT scan) |
+ |
– |
|
Relation of symptom duration and tumor size |
Large tumor with short duration of the symptoms |
Small tumor with long duration of the symptoms |
|
Score of -8 to+2: rather suspicious for carcinoma ->incision biopsy. Score of+3 to+8: suspicious for pleomorphic adenoma ->tumor resection in toto (incision biopsy is contraindicated). |
7.2.1.1.1.2 Warthin’s tumor (cystadenolymphoma) [245] [246]
-
One of the monomorphic adenomas
-
synonyms: adenolymphoma, cystadenolymphoma, papillary cystadenoma lymphatosum (however, to avoid confusion with malignant lymphomas or lymphadenomas, the term ‘Warthin’s tumor’ should be used)
-
-
Epidemiology
-
second most frequent benign tumor of the parotid gland, sometimes also found in periparotid lymph nodes
-
rarely in other salivary glands
-
extremely rare in the lacrimal gland (until now, fewer than 10 cases described worldwide)
-
-
Therapy
-
complete surgical resection via lateral orbitotomy
-
7.2.1.1.1.3 Oncocytoma [247] [248] [249] [250] [251] [252] [253] [254]
-
Synonyms
-
oxyphilic adenoma, oncocytic adenoma, Hürthle cell adenoma
-
-
Epidemiology
-
extremely rare in the lacrimal gland (MEDLINE analysis of the literature between 1959 and 2004 provided only 5 well-documented cases [248])
-
more frequently in salivary glands, kidney, adenohypophysis, (para)thyroid gland, caruncle, and conjunctiva of the eye
-
affects older women especially
-
-
Etiology
-
development probably from a mutation in the mitochondrial DNA
-
-
Histology
-
benign, slowly growing tumor originating from the epithelia of the excretory glandular ducts; malignant transformation extremely rare
-
granulated, large, eosinophilic, mitochondria-rich tumor cells, so-called oncocytes (“Hürthle cells”)
-
-
Therapy of choice
-
complete surgical resection
-
7.2.1.1.1.4 Myoepithelioma [241] [255] [256] [257] [258] [259]
-
One of the monomorphic adenomas
-
Epidemiology
-
Imaging
-
on CT, well-delimited encapsulated mass
-
in other locations, difficult to differentiate from other soft tissue tumors or sarcomas by clinical radiology
-
-
Therapy
-
complete and intact resection via orbitotomy, if needed, combined with coronal incision [255]
-
-
Follow-up
-
radiological follow-up recommended
-
-
Prognosis
-
local recurrence rate of 20%
-
7.2.1.1.1.5 Cystadenoma [260] [261] [262] [263]
-
Epidemiology
-
rare in the lacrimal gland
-
mainly found in the bile duct, pancreas, ovaries, epididymides, and kidneys
-
rare in the salivary glands (<1% of the salivary gland tumors)
-
-
Pathology
-
papillary and mucinous subtypes (the latter may degenerate)
-
first report in 2002 describing a bilateral cystadenoma of the lacrimal glands [260]
-
-
Differential diagnosis
-
papillary cystadenoma histologically similar to the papillary cystadenoma lymphomatosum (i. e., Warthin’s tumor)
-
7.2.1.1.1.6 Sclerotic polycystic adenosis (SPA) [264] [265]
-
Definition
-
new entity in the current WHO classification of epithelial salivary gland tumors
-
formerly considered as inflammatory/reactive change, current term ‘neoplasm’ controversially discussed but favored by most authors
-
-
Synonym
-
sclerotic polycystic adenoma
-
-
Epidemiology
-
first case report of SPA of the lacrimal gland in 2013 [264]
-
60 cases described to date (involvement mostly of the parotid gland, sometimes of the minor salivary glands)
-
-
Differential diagnoses
-
pleomorphic adenoma, adenoidcystic carcinoma, mucoepidermoid carcinoma, acinic cell carcinoma (often histopathological misinterpretation and confusion)
-
-
Prognosis
-
recurrence in 30% of cases
-
no metastatic spread or disease-related increased mortality because of SPA
-
#
7.2.1.1.2 Benign non-epithelial neoplasms of the lacrimal duct
7.2.1.1.2.1 Mesenchymal tumors
Solitary fibrous tumor {266–271]
-
Epidemiology
-
quite rare overall, regardless of location
-
2.8 cases per 100,000 patients in a Mayo Clinic population [268]
-
about 10 case reports of lacrimal gland involvement
-
typical locations: most frequently the pleura, more rarely abdomen, pelvis
-
-
Pathology
-
in the area of the lacrimal glands, possibly originating from periductal connective tissue
-
mostly benign, but malignant appearance (10–37%) as well as recurrences and metastases observed
-
slow, displacing growth
-
-
Differential diagnosis
-
in the area of the lacrimal gland, similar clinical and radiological appearance to pleomorphic adenoma
-
-
Therapy
-
complete resection via lateral orbitotomy
-
no clear evidence for benefit of radiotherapy or chemotherapy in cases of residual tumor
-
-
Prognosis
-
correlation between confirmed microscopically non–tumor-free surgical margins and increased development of local recurrences and distant metastases
-
local recurrences and distant metastases described mainly for tumors>10 cm, largely in the thorax or abdomen
-
extra-thoracic location linked to increased risk for local recurrences but not to the development of distant metastases
-
7.2.1.1.2.1.1 Myxoma [272] [273] [274] [275] [276]
-
Definition
-
neoplasm of mesenchymal/connective tissue origin
-
-
Epidemiology
-
1 case report involving the lacrimal gland
-
other locations at the eye: conjunctiva, cornea, orbit
-
other locations: predominantly heart, and also bones, skin, skeletal muscles, urogenital tract
-
-
Pathology
-
histologically few cells and vessels with abundant myxoid (mucus-like) matrix rich in hyaluronic acid
-
-
Diagnostics
-
well-circumscribed oval isodense mass on CT
-
-
Therapy
-
complete surgical resection (with excellent prognosis)
-
radiotherapy without relevant effect
-
7.2.1.1.2.1.2 Fibrous histiocytoma [261] [277]
-
Epidemiology
-
report on fibrous histiocytoma of the lacrimal gland in an 11-year-old girl, no other published case reports [277]
-
more frequent in the orbit of adults
-
-
Pathology
-
classification of benign, locally aggressive, and malignant types
-
7.2.1.1.2.2 Benign lymphoid tumors – reactive lymphoid hyperplasia [278]
-
Definition
-
benign lymphoproliferative disease
-
not a “true” neoplasm, being neither monoclonal nor autonomous
-
polyclonal lymphocytic proliferation
-
formerly called pseudolymphoma
-
atypical lymphoid hyperplasia as special type (borderline lesion between reactive lymphoid hyperplasia and lymphoma; today mostly classified as low-grade B cell lymphoma)
-
-
Epidemiology
-
about 6% of all lesions of the lacrimal gland [279]
-
similar incidence in conjunctiva, lacrimal glands, and other locations of the orbit (mostly extraconal), rarely in the eyelid
-
rarely bilateral or additionally outside the eye region (e. g., parotid, lung)
-
-
Clinical
-
slowly growing mass with minor functional impairment, only rarely reddening or pain
-
in the lacrimal gland, sometimes palpable as a rather solid, elastic mass with smooth or nodulous surface
-
-
Therapy [237]
-
high-dose glucocorticoids and/or radiotherapy (about 25 Gy)
-
rituximab for therapy-resistant cases
-
-
Prognosis
-
malignant transformation possible
-
#
#
7.2.1.2 Malignant neoplasms of the lacrimal gland
[Table 5] summarizes the malignant neoplasms of the lacrimal gland.
|
Tissue type |
Neoplasm |
|---|---|
|
Epithelial |
Adenoidcystic carcinoma |
|
Non-epithelial |
Synovial sarcoma |
|
Lymphoid |
Extranodal marginal zone B cell lymphoma |
7.2.1.2.1.Malignant epithelial neoplasms of the lacrimal gland
Comparing all types of carcinomas of the lacrimal and the major salivary glands, tumors with similar histology seem to have a less favorable prognosis when they appear in the lacrimal glands. For carcinomas of the lacrimal gland, a classification exists in the current TNM classification of malignant tumors of the UICC. According to this classification, preauricular, submandibular, and cervical lymph nodes are considered as regional lymph nodes. Assignment to the T categories T1–3 is based on tumor size; depending on the involvement of the periosteum and/or bone, the subcategories a–c are differentiated. An involvement of neighboring structures, e. g., paranasal sinuses, cavernous sinus, or brain, leads to categorization as T4 [280].
A singular histopathological classification for lacrimal gland tumors does not exist. Instead, many authors consider the lacrimal gland as a minor salivary gland and classify tumors based on the system for salivary glands.
[Table 6] shows a proposal for a classification of malignant epithelial lacrimal gland tumors, modified according to Weis et al., in analogy to the WHO classification of salivary gland tumors and with updates of the current version [237] [261] [285] [286]. It is worth mentioning that still some years ago primary adenocarcinomas of the lacrimal as well as salivary glands were not further subclassified. Meanwhile, it is known that they form a group of tumors with different morphologies and biological behaviors, and hence with different prognoses.
|
Classification |
Neoplasm |
|---|---|
|
Low-grade |
Carcinoma in pleomorphic adenoma (minimally
invasive carcinoma [≤1.5
mm]) |
|
High-grade |
Carcinoma (adenocarcinoma or adenoidcystic
carcinoma) in pleomorphic adenoma (minimally
invasive carcinoma [>1.5 mm]),
“malignant mixed
tumor” |
7.2.1.2.1.1 Adenoid cystic carcinoma (ACC) [279] [287] [288] [289] [290] [291] [292] [293] [294] [295]
-
Epidemiology
-
Pathology
-
perineural invasion of the tumor as a typical sign
-
frequently hematogenous metastatic spread (more typical than regional lymph node metastasis), even after several years (mainly in the lung, besides bones, liver, and brain)
-
-
Clinical
-
bulbus dislocation, proptosis, S-shaped ptosis, diplopia, epiphora
-
pain (in 38–79% of cases) and hypesthesia in the frontotemporal area (indicating the presence of an aggressive tumor)
-
symptom duration at first presentation typically<6 months
-
-
Diagnostics
-
high-resolution CT scan: bone erosion, irregular margins of the mass, possibly focal calcifications within the tumor
-
MRI: best option to identify involvement of the cavernous sinus, brain, and bone marrow
-
-
Therapy
-
still no clarity and no consensus regarding optimal therapy; mostly resection with safety margins and adjuvant radiotherapy
-
surgical options
-
according to extension, local resection, exenteration, or radical exenteration (with resection of the orbital roof, lateral orbital wall, and parts of the temporal muscle)
-
radical surgery without clear advantage for local tumor control up to T2, exenteration in cases of tumors at T3 or greater appears beneficial [297]
-
(elective) neck dissection: the rate of regional lymph node metastases (including the intraparotid lymph nodes) appears low in ACC (especially of the lacrimal gland) [297, 298); elective neck dissection appears unjustified in most cases (but should be discussed in advanced stages, solid histological subtypes, or high-grade malignant degeneration) [298]; higher lymphogenic metastasis rate with ACC of the salivary glands, with elective neck dissection thus leading to longer survival in advanced stages [299].
-
-
radiotherapy
-
in some trials, no significant survival benefit with additional radiation [288]; improved local control after adjuvant radiotherapy in larger tumors, per other studies [297]
-
particle therapy: satisfactory outcomes in ACC of the lacrimal gland with proton and heavy ion radiation [300] [301] [302] [303]
-
brachytherapy may be part of the therapeutic concept, e. g., for treatment of patients after R1 resection [304] [305]
-
-
chemotherapy: hints at improved prognosis with intraarterial cytoreductive chemotherapy (IACC) before (and after) therapy by means of surgery and radiation [289] [290]; furthermore, confirmed higher rate of eye-preserving surgeries after neoadjuvant IACC [306]
-
-
Prognosis
-
high rate of local recurrence
-
poor long-term prognosis: median survival of 5 years, 10-year survival rate of 20%
-
ACC of the lacrimal gland mainly involves high-grade phenotypes with aggressive behavior and shorter median survival (2.5 years) [297]
-
often death because of intracranial tumor growth or pulmonary metastases
-
7.2.1.2.1.2 Carcinoma in pleomorphic adenoma [252] [261]
-
Definition
-
according to the WHO classification of salivary gland tumors of 2017, the term ‘carcinoma in pleomorphic adenoma’ is only used together with the histological subtype [265]
-
-
Synonyms
-
pleomorphic (adeno-)carcinoma, malignant mixed tumor (misleading because only the epithelial (carcinoma) and not the mesenchymal part degenerates; different situation for carcinosarcoma (“true” malignant mixed tumor), also described in the lacrimal gland [282]
-
-
Epidemiology
-
second most frequent malignant epithelial tumor of the lacrimal gland (4–18%) [279] [291] [307] [308] [309]
-
appears typically in the 6th/7th decades of life (about 10 years later than pleomorphic adenoma)
-
adenocarcinomas more frequently observed in males, adenoid-cystic carcinomas more frequent in females [287]
-
-
Classification
-
non-invasive (intracapsular relation to pleomorphic adenoma)
-
minimally invasive (<4–6 mm above the capsula)
-
invasive (>4–6 mm above the capsula) [265]
-
-
Histology
-
Clinical
-
sudden development of tumor recurrence after incomplete removal of a pleomorphic adenoma or significant sudden progress (possibly with accompanying pain) of a tumor that has been symptom-free for several years
-
-
Therapy
-
Prognosis
-
mostly considered unfavorable but depends on several factors such as histological subtype and invasiveness
-
non-invasive and minimally invasive types: very good prognosis after complete surgical resection (without adjuvant therapy)
-
invasive type: very aggressive tumors with poor prognosis but favorable prognostic effect of adjuvant therapy [312] [313]
-
causes of death: intracranial spread, distant metastases (lung, thoracic wall, bones)
-
7.2.1.2.1.3 Adenocarcinoma (not otherwise specified, NOS) [233]
Annotation: In the current WHO classification of salivary gland tumors, the number of entities was reduced from 39 to 33 to reduce complexity and make the classification clearer. Among others, this change led to the inclusion of several, sometimes extremely rare entities under the term “adenocarcinoma NOS” [285].
-
Epidemiology
-
Etiology
-
de novo or within pleomorphic adenoma
-
-
Pathology
-
highly malignant
-
earlier metastasis comparison to adenoidcystic carcinomas
-
early lymphatic and hematogenous metastasis, mainly in the lung, brain, and mediastinum [314]
-
-
Therapy
-
frequently in an advanced stage at first presentation so that adequate surgical resection is difficult or even impossible
-
surgical therapy, if needed with orbital exenteration or craniofacial orbitectomy and regional lymph node dissection [291] [315]
-
with observed Her-2 overexpression in some adenocarcinomas of the lacrimal gland, pharmaceuticals such as lapatinib are possible therapeutic options [317]
-
-
Prognosis
-
shorter survival rates in comparison to adenoidcystic carcinoma
-
7.2.1.2.1.4 Mucoepidermoid carcinoma
-
Epidemiology
-
Clinical
-
typically indolent, slowly growing mass
-
-
Differential diagnosis
-
Therapy
-
Prognosis [314]
-
tumor-free survival and overall prognosis mainly grading dependent (classification into grades I–III based on histopathological characteristics)
-
Thorvaldsson et al. (1970): mean follow-up of 12 years in cases of mucoepidermoid carcinomas of the major salivary glands; survival rates of 100% or 97% in patients with grade I or grade II tumors, but only 43% in grade III tumors (high-grade) [314] [321]
-
7.2.1.2.1.5 Ductal adenocarcinoma
-
Epidemiology
-
Etiology
-
possible development as carcinoma in pleomorphic adenoma [324]
-
-
Pathology
-
-
therapy recommendations along the same lines as for ductal salivary gland carcinoma
-
surgical resection, if needed with adjuvant radio(chemo)therapy
-
orbital exenteration in>50% of the patients undergoing surgery
-
primary radiochemotherapy in non-resectable tumors
-
chemotherapy in palliative settings with response rates of 15–20%
-
if needed, pharmaceutical androgen deprivation therapy in the recurrent or metastatic setting, e. g., bicalutamide for androgen receptor–positive tumors or trastuzumab or lapatinib with Her-2/neu expression
-
-
Prognosis
7.2.1.2.1.6 Acinar cell carcinoma [328] [329] [330] [331]
-
Epidemiology
-
very rare in the area of the salivary glands (2–4% of parotid tumors)
-
even more rare in the area of the lacrimal gland, with only single case reports in the literature
-
females appear more frequently affected
-
age of manifestation: 6th decade of life
-
-
Diagnostics/differential diagnosis
-
uncharacteristic in CT/MR imaging, resulting in a high risk of confusion with pleomorphic adenoma
-
-
Therapy
-
surgical resection, eye preservation depending on the extent of the lesion, if needed, exenteration or advanced exenteration (in cases of bone infiltration)
-
7.2.1.2.1.7 Primary sebaceous carcinoma [332] [333] [334] [335]
-
Epidemiology
-
extremely rare, only single case reports
-
-
Hypotheses of pathogenesis
-
originating from heterotopic tissue
-
malignant transformation and sebaceous-like differentiation of other epithelial tumors of the lacrimal gland
-
originating from degenerated pluripotent cells
-
-
Pathology
-
highly malignant tumor often identified very late (high-grade)
-
-
Differential diagnoses
-
must be differentiated from secondary invasion (e. g., originating from upper eyelid carcinoma) and metastasis
-
histologically sometimes difficult to delimit from other entities such as squamous cell carcinoma or basal cell carcinoma (40–75% of sebaceous carcinomas of the lacrimal gland confused initially with squamous cell carcinoma; special staining for fat can avoid this confusion)
-
-
Therapy
-
surgical therapy, if needed, orbital exenteration or advanced exenteration
-
parotidectomy/neck dissection depending on the extension (infiltration of surrounding structures, e. g., the upper eyelid)
-
adjuvant radiotherapy (local recurrence rate after resection: 9–36%)
-
radiotherapy alone in cases of non-resectable tumors
-
particle therapy (e. g., heavy ions, C12) [336]
-
7.2.1.2.1.8 Myoepithelial carcinoma [337] [338] [339] [340] [341]
-
Epidemiology
-
Pathology
-
Therapy
-
overall, so rare that no therapy concept established
-
surgical therapy
-
radiotherapy
-
applied in myoepithelial tumors of the lacrimal gland at least in the palliative setting [344]; if the parotid gland or other locations of the head and neck involved, adjuvant therapy after surgical resection in half of cases [341]
-
primary radiotherapy if surgical resection not possible
-
significance of adjuvant radiation unknown; several authors report no effect on local recurrence rate [345]
-
-
chemotherapeutics partly described as effective, but systematic data are lacking
-
-
Prognosis
-
high-grade malignancy with poor prognosis and high recurrence rate [286] [341]
-
myoepithelial carcinoma of the head and neck: 5-year survival rate of 32% [341]
-
distant metastasis rate of 35% (mainly pulmonary metastases) [341]
-
distant metastases with primary tumor in the lacrimal gland rarely described (cave: low number of cases) [342]
-
7.2.1.2.1.9 Squamous cell carcinoma
-
Epidemiology
-
according to a review by Weis et al. (2009), only 8 case reports published at the time of their writing [286]
-
-
Etiology/pathogenesis
-
Therapy
7.2.1.2.1.10 Oncocytic carcinoma [349] [350]
-
Epidemiology
-
according to Kalantzis et al. (2013), at the time of their writing, four published cases with involvement of the lacrimal gland [350]
-
-
Pathology
-
high-grade tumor with infiltrative growth pattern, tendency to local recurrences and distant metastases
-
-
Therapy
-
radical resection (if needed, orbital exenteration) followed by adjuvant radio(chemo)therapy
-
7.2.1.2.1.11 Polymorphic adenocarcinoma [265] [351]
-
Formerly used term (until 2017)
-
polymorphic low-grade adenocarcinoma
-
-
Epidemiology
-
according to Selva et al. (2004), at the time of their writing, four published cases with involvement of the lacrimal gland [351]
-
appears almost exclusively in the minor salivary glands, mainly at the palate (female:male ratio, 2:1)
-
-
Differential diagnosis
-
often confused with adenoidcystic carcinoma
-
-
Pathology
-
infiltrative growth and perineural invasion
-
metastases in regional lymph nodes relatively rare (6.5–10%), distant metastases extremely rare
-
-
Therapy
-
Prognosis
-
clearly better compared to other adenocarcinomas of the salivary gland (overall survival of ≥95%)
-
if salivary glands involved, local recurrence in 5–33% of cases (generally well controllable with re-resection)
-
7.2.1.2.1.12 Secretory carcinoma [265] [352] [353]
-
Definition
-
new entity, first described in 2010 in the head and neck as mammary analogue secretory carcinoma, designated as “secretory carcinoma” by the WHO
-
previously assigned to other entities despite not always typical histopathological characteristics (e. g., granule poor acinic cell carcinoma)
-
-
Pathology
-
similar to the secretory carcinoma of the breast, also identification of the specific ETV6-NTRAK3 fusion gene
-
occurring in the head and neck mainly in the parotid gland (70%), and also in the buccal, lip, and palatal mucosa, rarely in the submandibular or sublingual glands
-
regional lymph node metastases in up to 25% of cases
-
-
Therapy
-
surgical resection
-
tropomyosin receptor kinase (TRK) inhibitors (larotrectinib, entrectinib) as future therapy options for tumors with NTRK fusion
-
-
Prognosis
-
overall, not very aggressive tumor with relatively favorable prognosis (survival exceeding 95%)
-
7.2.1.2.2 Malignant non-epithelial tumors of the lacrimal gland
Malignant non-epithelial tumors of the lacrimal gland are summarized in [Table 7]. Lymphomas of the lacrimal gland are discussed specifically in section 7.2.1.2.3.
|
Neoplasm |
Characteristics |
|---|---|
|
Synovial sarcoma [354] |
|
|
|
|
Malignant peripheral nerve sheath tumor [357] [358] (terms that are synonymously used such as neurogenic sarcoma, neurofibrosarcoma, and malignant schwannoma should be avoided) |
|
|
7.2.1.2.3 Malignant lymphoproliferative diseases – lymphomas of the lacrimal gland [233] [237] [365] [366]
-
Definition
-
belongs to ocular adnexa lymphomas
-
-
Epidemiology
-
Classification: by decreasing incidence, the following entities are found in the lacrimal gland
-
extranodal marginal zone lymphoma
-
follicular lymphoma
-
diffuse large cell B cell lymphoma (DLBCL)
-
-
Etiology/pathogenesis
-
in rare cases, development on the floor with chronic inflammatory diseases (Sjögren’s syndrome, IgG4-associated diseases, reactive lymphatic hyperplasia)
-
-
Clinical
-
Diagnostics
-
imaging procedures
-
CT scan or MRI: smooth and homogeneous mass, mostly without infiltration of the surrounding tissue (but possible bone erosion in highly aggressive types [365])
-
clear morphological differentiation among reactive lymphatic hyperplasia, lymphoma, and non-specific inflammation of the lacrimal gland not possible with clinical assessment or imaging, so biopsy required (incision biopsy preferable to fine needle aspiration cytology) [233]
-
-
further diagnostics (for assessment of systemic involvement)
-
bone marrow biopsy, whole body CT scan/MRI or PET
-
Ann Arbor classification and TNM staging apply
-
-
-
Therapy
-
radiotherapy, especially in localized types (Ann Arbor stage IE)
-
polychemotherapy, especially in cases of systemic spread (stages IIE-IVE), in cases of DLBCL also in an early stage [369]
-
antibody therapy, e. g., anti-CD-20 rituximab
-
stem cell transplantation, if needed
-
surgical therapy: surgical therapy alone generally not justified [233]
-
-
Prognosis
-
at the time of first diagnosis, about 80% at a localized stage (IE), and 20% showing systemic involvement in the context of staging (stage IIE–IVE) [233]
-
for patients with stage IE at first diagnosis, secondary generalization in the further course in 22–42% of cases [233]
-
Rasmussen et al. (2011): 5-year survival rate of 70% in 27 patients with lymphoma of the lacrimal gland [366]
-
7.2.1.2.3.1 Extranodal marginal zone B-cell lymphoma
-
Epidemiology
-
most frequent subtype in the lacrimal gland, with 40% of all lymphomas [366]
-
-
Therapy
-
early stage (stage IE/IIE, no systemic involvement): radiotherapy (about 30–40 Gy) [370]
-
in cases of systemic involvement or high tumor charge: pharmaceutical combination therapy, e. g., rituximab+cyclophosphamide+hydroxydaunorubicin+vincristine+prednisone (R-CHOP) ->3-year progression-free survival in 70–80%
-
rituximab applied for a further 2 years as maintenance dose
-
-
Prognosis
-
low-grade lymphoma: 5-year survival rate of 75% [366], more favorable in salivary glands (95–100%)
-
7.2.1.2.3.2 Follicular lymphoma
-
Epidemiology
-
Therapy
-
early stage (stage IE/IIE): radiotherapy ->5-year survival rate of about 100% [366]
-
in cases of systemic involvement or high tumor charge: pharmaceutical combination therapy, e. g., R-CHOP ->3-year progression-free survival in 70–80%
-
rituximab applied for a further 2 years as maintenance dose
-
-
Prognosis
-
low-grade lymphoma with favorable prognosis (5-year survival of about 80–90%)
-
7.2.1.2.3.3 Diffuse large-cell B cell lymphoma (DLBCL)
-
Epidemiology
-
Pathology
-
destructive growth in neighboring structures (e. g., ethmoid, intracranial) observed in 6 of 27 patients [365]
-
-
Therapy
-
already in stage I (only involvement of the lacrimal gland): R-CHOP
-
in patients younger than 60 years with high risk profile (IPI score≥2 or increased LDH), etoposide added to R-CHOP (R-CHOEP)
-
in stage IE and absence of B symptoms, additional radiotherapy
-
-
Prognosis
-
high-grade lymphoma; 5-year survival rate: 55–73%
-
7.2.1.2.3.4 Mantle cell lymphoma
-
Therapy
-
primary therapy: R-CHOP
-
in patients younger than 65 years, additional cytarabine and stem cell transplantation
-
-
Unfavorable prognosis
Annotation: The WHO classification of lymphoid neoplasms encompasses more than 80 lymphoma categories. They are subdivided into three groups: B cell neoplasms, T and NK cell neoplasms, and Hodgkin lymphomas. Lymphomas as well as lymphoid leukemia are included. At this point, only the most important entities are mentioned [371]. The incidence distribution of the subtypes in the lacrimal gland corresponds to that for the salivary glands [366].
#
7.2.1.3 Metastases in the lacrimal gland
-
Epidemiology
#
#
7.2.2 Neoplasms of the orbit (without lacrimal gland neoplasms)
7.2.2.1 Benign neoplasms
7.2.2.1.1 Benign epithelial neoplasms
See Section 6.2.1.1.1, “Benign epithelial neoplasms of the lacrimal gland.”
#
7.2.2.1.2 Benign non-epithelial neoplasms
Tumors of non-epithelial origin are even more numerous in type than epithelial tumors of the orbit.
#
7.2.2.1.2.1 Fibro-osseous processes of the orbit
There are several fibro-osseous lesions that can appear in the periorbital region and may lead secondarily to involvement of the orbital soft tissue, including an aneurysmal bone cyst, giant cell granuloma, and cholesterol granuloma. However, in the literature, these lesions are often called ‘reactive bone lesions’ and included in the list of bone tumors of the orbit.
7.2.2.1.2.1.1 Fibroma, ossified fibroma, fibromatosis [201] [373]
Fibroma
-
Epidemiology
-
Pathology
-
mostly originating from the cranial periorbita or muscle conus
Ossified fibroma
-
-
Epidemiology
-
occurring mainly in younger patients
-
-
Pathology
-
slowly growing benign tumors, but destructive growth and recurrence possible after incomplete resection
-
location also in the paranasal sinuses of maxillary bones
-
-
Diagnostics
-
radiological differentiation from fibrous dysplasia necessary
Fibromatosis
-
-
May have an intermediate character
-
Therapy
-
generally surgical resection
-
7.2.2.1.2.1.2 Aneurysmal bone cyst

-
Definition
-
locally expansive osteolytic lesion consisting of blood-filled cavities with surrounding reactive fibro-osseous tissue
-
-
Location
-
Epidemiology
-
preferred occurrence in childhood and adolescence up to the 20th year of life
-
-
Etiology and pathogenesis
-
association with other bone lesions (fibrous dysplasia, chondroblastoma, osteoblastoma, giant cell tumors) that precede development of aneurysmal bone cyst and may be predisposing for the development as a reactive phenomenon [382] [383]
-
focal hemodynamic changes with secondary venous hypertonia or preexisting trauma discussed as causes [381]
-
current oncogenetic study results: clonal neoplastic lesion with specific genetic rearrangement (USP6 gene upregulation in spindle cells) [384], leading to possible differentiation of similar lesions without USP rearrangement (e. g., osteosarcoma, chondroblastoma, giant cell tumors)
-
-
Clinical
-
initially indolent swelling or bulbar protrusion
-
often slow growth over weeks and months, then episodes of rapid growth and painful exophthalmos, ptosis, diplopia, and further ophthalmological symptoms [378]
-
-
Diagnostics
-
CT scan and MRI (characteristic changes, see [Fig. 16a, b]): polylobulated mass with multiple cystic areas, separated by connective tissue septa; furthermore, solid areas, thinning of the surrounding cortex, liquid level in the cystic areas because of non-coagulated blood; clear contrast enhancement, the dura respected in cases of intracranial extension [378] [385]
-
histology: cystic cavities lined with endothelial-like cells; solid parts consisting of spindle-shaped fibroblasts with singular multinucleated giant cells, hemosiderin-charged macrophages, granulation tissue, and extravasal erythrocytes; septa frequently with newly developed bone; no atypical mitoses or other histological malignancy criteria
-
-
Differential diagnoses
-
other bone tumors (e. g., osteoblastomas, osteoclastomas, brown tumors)
-
reparative giant cell granuloma and giant cell tumors
-
examination by pathology reference centers required [378]
-
-
Therapy
-
first choice: surgical excision and (depending on the extension and local destruction) reconstruction of the bone defect
-
in extended cases: intralesional curettage and partial removal with adjuvant radiotherapy
-
in cases of high intraoperative risk of bleeding: preoperative embolization or stereotactic radiosurgery
-
-
Prognosis
7.2.2.1.2.1.3 Giant cell granuloma [387] [388] [389] [390] [391]
-
Definition
-
rare, reactive benign mass with locally aggressive growth
-
-
Epidemiology
-
mostly in females in the 1st to 3rd decades [392]
-
-
Location
-
Pathogenesis
-
still unknown
-
inflammation, trauma, and intraosseous hemorrhages are possible causes
-
because of the association with other symptoms such as Noonan syndrome, neurofibromatosis type I, and cherubism, a still unidentified chromosomal anomaly appears possible [393]
-
-
Clinical
-
symptoms mostly the result of processes involving masses: swelling, deformity, sometimes pain
-
exophthalmos, visual impairment, diplopia as first clinical symptoms
-
-
Diagnostics
-
CT scan: mostly sharply delimited osteolytic process with expansive character, but highly variable and non-specific
-
histopathology: characteristic accumulation of multinucleated large osteoclast-like giant cells with a tendency to cluster around hemorrhagic foci
-
-
Differential diagnoses
-
osteoclastoma, osteosarcoma, eosinophilic granuloma, aneurysmal bone cyst, “brown tumors” in cases of hyperparathyroidism, non-ossified fibroma, Langerhans cell histiocytosis (radiological differentiation sometimes difficult)
-
differentiation of giant cell tumors is controversially discussed in the literature [394]: in the described cases, definite diagnosis of orbital giant cell granuloma was only possible by biopsy or surgical excision
-
-
Therapy
-
first choice: complete resection
-
not always possible as radical intervention in the area of the skull
-
radiation: controversially discussed in cases of resistance to radiation and possible malignant cell degeneration; however, recommended by some authors after partial resection
-
medication for adjuvant therapy: corticosteroids, calcitonin, bisphosphonates, imatinib, and interferon-α [393] [395]
-
denosumab (monoclonal antibody): neutralization by binding to the RANK ligands produced by the tumor cells in cases of unresectable or locally advanced findings [391]
-
-
Prognosis
-
recurrence in 10–50% of cases
-
7.2.2.1.2.1.4 Cholesterol granuloma
-
Definition
-
rare benign lesion developing because of foreign body reaction to cholesterol crystals
-
-
Epidemiology
-
occurring typically in middle-aged males [396]
-
-
Etiology and pathogenesis
-
presumed development from traumatic genesis or intrinsic anomaly of diplopia
-
inflammatory reaction against degradation products of blood components or in cases of hypoxic tissue destruction
-
granulomatous inflammatory reaction on cholesterol with subsequent erosion of the surrounding bone [397] [398]
-
-
Location
-
mostly in the temporal bone
-
possible also in the area of the facial skull
-
rarely orbital, but if so, typically in the area of the frontal bone above the lacrimal fossa (superotemporal or superior) because the weakest connection of the periorbita to the bone is found with frequent occurrence of trauma [399] [400] [401]; in cases of unclear lesions of the orbit from imaging, location, and often positive trauma history, cholesterol granuloma to be considered [401]
-
-
Clinical
-
exophthalmos, diplopia, visual impairment
-
more rarely periorbital pain or ptosis
-
-
Diagnostics
-
CT scan: osteolytic lesions of the frontal diploe with erosion of the adjacent cortex and sometimes expansion into the extradural space
-
MRI: differentiation of other soft tissue tumors is possible; typically high signal intensities in the T1- and T2-weighted sequences because of liquid content [402]
-
histology: cholesterol crystals surrounded by multinucleated giant cells; differentiation of epidermoids and dermoid cysts because of absence of epithelial elements; differentiation of aneurysmal bone cyst or giant cell tumors by the prominent xanthomatous component
-
-
Differential diagnoses
-
difficult clinical and CT morphological differentiation of dermoid or epidermoid cyst
-
others: neoplasms of the lacrimal gland, metastases, aneurysmal bone cyst, or chondroma [61]
-
-
Therapy
7.2.2.1.2.1.5 Osteoma [201]
-
Epidemiology
-
most frequently a tumor originating from the orbital bone (often also from the paranasal sinuses)
-
-
Diagnostics
-
CT scan: bone-dense, well-delimited mass (often incidental finding)
-
-
Therapy
-
surgical resection, only in symptomatic/progressive cases
-
7.2.2.1.2.1.6 Chondroma [201] [404]
Annotation: The trochlea is the only cartilaginous structure of the orbit, and the sphenoid has cartilaginous precursor structures.
-
Epidemiology
-
Garrity and Henderson (2007) [405] and Shields et al. (2004) [279]: only one case each out of 1373 and 627, respectively, of mesenchymal orbital tumors
-
Rootman et al. (2004): one case out of 62 primary tumors of the orbital bone in a period of 24 years [405] [406] [407]
-
in the area of the facial skull, mainly occurring in adolescents and young adults
-
-
Clinical
-
often asymptomatic
-
in cases of progression, ptosis/proptosis
-
-
Location
-
superior nasal quadrant
-
-
Pathology
-
histopathological differentiation of low-grade chondrosarcomas is difficult
-
partly aggressive behavior of chondrogenic tumors of the facial skeleton (discrepancy between histological appearance and biological behavior)
-
-
Therapy
-
resection, with a safety margin if possible
-
long-term follow-up recommended [404]
-
7.2.2.1.2.2 Neuroma [201] [408]
-
Synonyms
-
schwannoma, Schwann cell tumor
-
-
Epidemiology
-
0.7–2.3% of orbital tumors
-
usually singular occurrence
-
in up to 18% of cases, association with neurofibromatosis
-
-
Pathology
-
benign tumor of the peripheral nerves
-
-
Therapy
-
surgical resection, especially in symptomatic processes
-
7.2.2.1.2.3 Neurofibroma [201] [409]
Plexiform neurofibroma
-
Epidemiology
-
most frequent subtype in the orbit
-
often congenital or in early childhood
-
neurofibromatosis type I is often the primary disease
-
-
Pathology
-
multinodular mass with infiltrative behavior (eyelid, skull base, periorbita) and relatively strong vascularization
-
degeneration rate (transformation into malignant peripheral nerve sheath tumor): 2–4%
-
-
Clinical
-
severe visual impairment is typical
-
-
Therapy
-
surgical resection
-
difficult because of irregular borders, often multiple intervention until exenteration
-
-
Epidemiology
-
rarely associated with neurofibromatosis
-
manifestation peak: 3rd-5th decades of life
-
-
Location and pathology
-
typically cranial location in the orbit (frontal nerve), less vascularized, better encapsulated
-
-
Therapy
-
surgical resection
-
-
Pathology
-
in contrast to schwannomas, additionally fibroblasts and perineural cells are found, higher contents of connective tissue substance, not (well-)encapsulated, cannot be separated surgically from the nerve, requiring sacrifice of the nerve
-
7.2.2.1.2.4 Meningioma [201] [411]
-
Epidemiology
-
according to estimations, about 4–8% of all orbital neoplasms
-
age of manifestation: typically around the 45th year of life, optic nerve sheath meningioma in younger patients
-
women significantly more frequently affected than men
-
-
Risk factors
-
previous radiotherapy of the head region
-
neurofibromatosis type 2
-
-
Typical locations
-
lateral orbital wall, orbital roof (“primary ectopic meningiomas,” only single cases)
-
optic nerve sheath meningioma (most frequent optic nerve sheath tumor), in 90% of cases, already growth through the optic nerve canal in direction of the chiasma at diagnosis [412]
-
ingrowth of meningiomas of the cavernous sinus or the medial sphenoid wing (optic nerve compression)
-
-
Pathology
-
orbital meningiomas usually grade 1 meningiomas according to the WHO grading for meningiomas (grade 1: benign; grade 2: anaplastic; grade 3: malignant) [413]
-
-
Clinical
-
leading symptoms
-
slowly progressive, painless loss of vision (key symptom)
-
visual field defects
-
transient obscurations
-
protrusion or motility disorders (with diplopia) often in advanced stage
-
-
-
Therapy [411] [412] [414] [415]
-
with small and/or less impairing findings: “watch-and-wait” and initially half-yearly MRI controls
-
therapy indication [412]: progressive visual impairment, visual decline to 0.4; increasing visual field disorders, endangered chiasma
-
optic nerve sheath meningioma
-
therapy of first choice: fractionated radiotherapy (50–55 Gy) with probably best functional outcome (vision)
-
surgical therapy, especially in cases of extension in intracranial direction or blindness and disfiguring protrusion
-
in cases of growth into the optic canal, optic nerve decompression, if needed
-
surgical resection (if necessary, embolization prior to surgery), possibly with adjuvant radiotherapy
-
-
7.2.2.1.2.5 Opticus glioma [201] [412]
-
Definition
-
special type of pilocystic astrocytoma (juvenile pilocystic astrocytoma, WHO grade I) [412]
-
-
Epidemiology
-
manifestation in 90% of cases before the 20th year of life, in 50% of cases before the age of 5 [412]
-
underlying neurofibromatosis type I possible and should be excluded
-
-
Differential diagnosis
-
optic nerve schwannoma [416] (observed, even though extremely rare [fewer than 10 cases have been described], even if the myelin sheath of the optic nerve does not contain Schwann cells; most probably originating from small sympathetic nerve fibers)
-
-
Clinical
-
bilateral location possible (especially in neurofibromatosis)
-
slow growth
-
exophthalmos
-
squint position of the affected eye
-
visual and visual field impairment
-
phase-wise increase of the complaints in cases of bleeding
-
nystagmus
-
endocrinological accompanying symptoms possible (e. g., diabetes insipidus, obesity, precocious puberty)
-
-
-
controversial because a spontaneous course is not predictable, spontaneous remission is possible, and all procedures have some significant side effects
-
proximity to the optic canal is crucial for the course
-
follow-up by means of MRI and visual/visual field examinations (more decisive for the therapy indication than radiologically found increase) in cases of newly diagnosed gliomas
-
(partial) resection, especially in cases of large, progressive intraorbital gliomas with blindness, growth in direction of the chiasma, or disfiguring exophthalmos
-
probably transection of the optic nerve in front of the chiasma to avoid extension to the contralateral side
-
fractionated radiotherapy was former therapy of choice, but in children, especially age<7 years, associated with the risk of neurological-cognitive deficits and later secondary tumors (in particular with neurofibromatosis), so currently indicated mainly in progressing chiasma-near tumors in older children or adults
-
chemotherapy to reduce tumor size and growth and to delay other therapies
-
first-line therapy in cases of loss of vision/tumors requiring action especially in small children: pharmaceuticals such as carboplatin combined with vincristine
Case report 3
-
-
History
-
18-year-old female patient
-
increasing exophthalmos for about 4 months and visual impairment for 5 days, left side
-
-
Diagnostics
-
ophthalmological examination
-
vision: right side 1.25, left side 0.6
-
Hertel exophthalmometry: right side 20, left side 24 mm
-
congestive papilla, left side
-
-
MRI ([Fig. 17])
-
suspected optic nerve glioma measuring 28×17 mm
-
no intracerebral growth
-
-
-
Procedure
-
transnasal endoscopic medial orbital decompression with biopsy
-
histopathology: optic nerve glioma
-
definitive fractionated stereotactic radiotherapy (50.4 Gy)
-
-
Course
-
initially fluctuating but good vision (around 0.8–0.9)
-
about 6 months after radiation: ophthalmological optic nerve atrophy, clearly deteriorated vision of the left eye (0.25), increasing edema of the papilla, secondary strabismus divergens, cosmetically disturbing progressive exophthalmos ([Fig. 18]), and insufficient eyelid closure
-
further deterioration during the following 3 months (vision 0.1, Hertel exophthalmometry on the left side 27 mm, increasing suffering, fundoscopy findings ([Fig. 19])
-
bulbus enucleation and tumor resection, implantation of Guthoff implant, later prosthesis
-
genetic assessment to exclude neurofibromatosis
-



7.2.2.1.2.6 Myxoma [272] [273] [274] [275] [276]
-
Definition
-
neoplasm of mesenchymal/connective tissue origin
-
-
Epidemiology
-
few case reports published about the orbital area
-
men and women equally affected
-
-
Pathology
-
histologically containing few cells and vessels with abundant myxoid (mucus-like) hyaluronic acid-rich matrix
-
other locations: predominantly heart, along with bones, skin, skeletal muscles, urogenital tract
-
-
Diagnostics
-
association of orbital myxomas with Carney syndrome (familial myxoma syndrome), Mazabraud syndrome, McCune-Albright syndrome
-
should be considered in patients with increased numbers of chloasma, cardiac history, and orbital mass
-
a well-circumscribed, oval, isodense mass seen on CT
-
radiotherapy without relevant effect
-
Further examples of benign non-epithelial orbital neoplasms are listed in [Table 8].
|
Neoplasm |
Characteristics |
|---|---|
|
Granular cell tumors [420] (Abrikossoff tumor) |
|
|
|
|
Ganglioneuroma [423] [424] (benign type of neuroblastic tumors) |
|
|
Ganglioneuroblastoma [423] (intermediary type of neuroblastic tumors) |
|
|
|
|
|
|
|
|
Melanotic neuroectodermal tumor of infancy (i. e., MNTI) [431] [432] [433]; former synonyms: melanotic ameloblastoma, retinal predisposed tumor, melanotic progonoma, melanotic adamantinoma |
|
#
#
7.2.2.2 Malignant neoplasms
7.2.2.2.1 Malignant epithelial neoplasms
See Section 7.2.2.2.3, “Malignant epithelial tumors of the lacrimal gland.”
#
7.2.2.2.2 Malignant non-epithelial neoplasms
Annotation: Lymphomas of the orbit are described in Section 6.2.2.2.3.
7.2.2.2.2.1 Rhabdomyosarcoma [434]
-
Epidemiology
-
most frequent orbital malignancy in children
-
embryonic subtype: most frequent, about 60–70% of all pediatric rhabdomyosarcomas
-
alveolar subtype: 30% of all pediatric rhabdomyosarcomas, in 80% associated with specific gene translocation/-fusion (PAX3 on chromosome 2 or PAX7 on chromosome 1 with FOXO1 on chromosome 13)
-
-
Clinical
-
rapidly progressing exophthalmos
-
eyelid swelling/reddening
-
ptosis, possibly palpable tumor
-
-
Diagnostics
-
biopsy
-
-
Therapy
-
standard: chemotherapy+radiotherapy
-
exenteration not to be performed primarily, only with confirmation of residual tumor/recurrence in the biopsy after chemotherapy+radiotherapy
-
-
Prognosis
-
key problem of local recurrence
-
unfavorable prognosis in cases of confirmed gene translocation/fusion
-
#
7.2.2.2.2.2 Fibrosarcoma [435] [436] [437]]
-
Epidemiology
-
mean age at manifestation: 50 years
-
men preferentially affected (independent of location)
-
several case reports of orbital involvement in children
-
-
Etiology and pathogenesis
-
occurrence as post-radiogenic secondary tumor in the orbit (e. g., after radiotherapy of retinoblastoma)
-
increased risk after implantation of foreign material
-
-
Pathology
-
aggressive tumor (in more than 80% grade 2/3 according to FNCLCC)
-
metastases mainly in the lung, bones, and regional lymph nodes
-
only single case reports of orbital fibrosarcomas after application of modern diagnostic procedures
-
in recent years, improved histopathological diagnostic procedures and classification systems for sarcomas have led to a rare diagnosis of exclusion (<1% of all adult soft tissue sarcomas)
-
-
Therapy
-
resection with wide safety margins up to exenteration
-
exenteration especially in cases of local recurrences
-
significance of postoperative radio- or chemotherapy unknown, in particular in cases of rare locations in the orbit
-
-
Prognosis
-
5-year survival rate of about 55%
-
#
7.2.2.2.2.3 Retinoblastoma [438] [439] [440]
-
Definition
-
malignant tumor in the retina; precondition for its development are mutations in both alleles of the retinoblastoma gene (located on chromosome 13, band q14)
-
-
Epidemiology
-
incidence of about 1:20 000 live births
-
most frequent intraocular malignancy in children
-
male:female ratio, 1:1
-
usually occurring before the age of 5 years
-
-
Types
-
45% inherited
-
heterozygous for mutations in the retinoblastoma gene, with an additional mutation arising in the second allele
-
both eyes usually affected
-
-
mean age at the time of diagnosis: 12 months
-
55% non-inherited
-
both mutations occurring in the same cell
-
mainly unilateral
-
mean age at the time of diagnosis: 23 months
-
-
75% unilateral, 25% bilateral
-
-
Clinical
-
leukocoria, strabismus, more rarely: painful reddening of the eye, glaucoma, visual impairment
-
-
Diagnostics
-
ophthalmological examination: unifocal or multifocal whitish, vascularized retinal mass, sometimes with tumor dissemination
-
ultrasound
-
MRI (for evaluation of optic nerve involvement and possible extraocular extension)
-
gene diagnostics (differentiation between hereditary and non-hereditary type)
-
generally no biopsy
-
lumbar puncture in cases of extension in direction of the CNS
-
possibly bone scintigraphy/bone marrow puncture if bone metastasis is suspected
-
-
Therapy
-
objective: preservation of life (primary objective), preservation of function (secondary objective)
-
laser/cryocoagulation, transpupillary thermotherapy (especially in small tumors)
-
chemotherapy as one of the most important options: allows avoiding radiotherapy or enucleation in a significant number of cases
-
enucleation including resection of the optic nerve (especially in advanced unilateral tumors)
-
radiotherapy (former standard therapy with high radiation sensitivity, but significant risk for post-radiogenic secondary tumors – about 36% within 50 years in the hereditary type – so reserved for specific indications such as metastatic spread or response failure on chemotherapy)
-
brachytherapy: primary therapy option in cases of solitary tumors ventral to the equator (in 2/3 of cases with good visual function) or after failure of other therapeutic strategies, adjuvant therapy option
-
adjuvant chemotherapy, especially after enucleation if certain risk factors are present (e. g., choroid invasion, optic nerve invasion, extension into the orbit, dissemination into the anterior chamber)
-
-
Prognosis
-
5-year survival rate: about 96% in developed countries
-
in the hereditary type: increased risk for secondary (extraorbital) tumors
-
#
7.2.2.2.2.4 Malignant glioma of the optic nerve [412] [441] [442]
-
Definition
-
corresponds to anaplastic astrocytoma, WHO grade III, or glioblastoma, WHO grade IV; no association with neurofibromatosis
-
-
Epidemiology
-
extremely rare
-
manifestation peak: middle-aged adults
-
-
Symptoms
-
initially similar to acute neuritis of the optic nerve
-
retroorbital pain
-
at time of diagnosis, chiasma involvement already present in 75%, early CNS infiltration
-
rapidly progressive loss of vision up to blindness
-
possibly other neurological symptoms such as seizures
-
-
-
no effective therapy
-
combined radiochemotherapy with temozolomide
-
surgical resection only in single cases, e. g., strictly unilateral involvement without chiasma involvement
-
-
Prognosis
-
death usually within one year
-
Further examples for non-epithelial malignant neoplasms of the orbit are listed in [Table 9].
|
Neoplasm |
Characteristics |
|---|---|
|
|
|
|
|
Ewing sarcoma [449] [450] (former: tumors of the Ewing sarcoma family [Ewing sarcoma of the bone (ESB), extraosseous Ewing sarcoma (EES), peripheral primitive neuroectodermal tumor (pPNET) of the bone, and Askin’s tumor of the chest wall]) |
|
|
|
|
|
|
Epitheloid sarcoma [456] |
|
|
Neuroblastoma [423] [457] [458] [459] (malignant type of neuroblastic tumors) |
|
|
Malignant peripheral nerve sheath tumor [412] |
|
|
Orbital melanoma [460] |
|
|
#
7.2.2.2.3 Lymphoproliferative diseases of the orbit
7.2.2.2.3.1 Orbital lymphomas [365] [463] [464] [465]
Annotation: See Section 7.2.1.2.3, “Lymphomas of the lacrimal gland.”
-
Epidemiology of lymphomas
-
the most frequent malignant orbital masses (about 50–55% of all orbital malignancies are non-Hodgkin lymphomas)
-
the most frequent primary orbital tumors in older patients (>60 years)
-
lymphoid tumors encompass about 10–20% of orbital masses
-
ocular adnexae (conjunctiva, eyelids, lacrimal gland, orbital soft tissue) affected in 1–2% of all lymphomas and about 8% of all extranodal lymphomas
-
secondary orbital involvement in an estimated 5% of all patients with non-Hodgkin lymphomas
-
mainly older people affected
-
-
Frequency in lymphoma types
-
B cell lymphomas, 97%
-
(extranodal) marginal zone (B cell) lymphomas, about 50–60%
-
follicular lymphoma, about 9–23%
-
DLBCL, about 8–23%
-
mantle cell lymphoma, about 5%
-
-
Locations
-
orbital soft tissue: 32–60%
-
conjunctiva: 15–42%
-
lacrimal gland: 14–20%
-
external eye muscles: 9%
-
lacrimal sac: 2–11%
-
bilateral occurrence: 11–32%
-
-
Clinical
-
lymphomas of the conjunctiva: typically smooth and salmon-colored
-
morphologically sometimes difficult to differentiate from idiopathic orbital inflammation by imaging (formerly so-called orbital pseudotumor) ->biopsy
-
mostly locally limited ocular adnexa lymphomas at the time of diagnosis (exception: mantle cell lymphomas)
-
-
Therapy
-
in localized manifestation: radiotherapy ->good local control and healing rate
-
in cases of bilateral or systemic disease and aggressive subtypes (e. g., DLBCL): chemotherapy
-
immunotherapy with anti-CD-20 antibodies
-
-
Prognosis
-
low-grade lymphoma (marginal zone lymphoma, follicular lymphoma): good
-
high-grade lymphoma (DLBCL, mantle cell lymphoma): poor
Case report 4
-
-
History
-
first presentation of a 58-year-old male patient in the Department of Ophthalmology of the University Hospital of Halle, Germany
-
complaints for 2 years
-
-
Symptoms
-
left eye protruded and reddened
-
current visual impairment
-
[Fig. 20a, b] shows preoperative findings
-
-
Previous measures and course
-
intensive diagnostics (including MRI examination) and therapy by ophthalmologists in cooperation with a general hospital
-
transconjunctival biopsy of the orbit performed elsewhere: on histopathology, a lymphoplasmacellular infiltrate without a conspicuous IgG4 ratio
-
working diagnosis: idiopathic inflammatory orbitopathy, left more than right
-
-
therapy-refractory course with high-dose glucocorticoids for about 1.5 years (80 mg prednisolone at the time of first presentation), obvious Cushing symptoms, steroid-induced diabetes mellitus, arterial hypertonia, NYHA III heart failure, liver steatosis
-
-
Diagnostics/clinical findings (relevant aspects)
-
no relative afferent pupillary defect, impaired bulbus motility (especially adduction) on the left, diplopia when looking upwards and to the side
-
anterior segments of the eye
-
congested conjunctival/episcleral vessels
-
lid-parallel conjunctival folds (LIPCOF)++
-
-
fundoscopy
-
left papilla temporally faint
-
retina and choroid folds
-
-
vision
-
right: 1.0
-
left: 0.4
-
-
Hertel exophthalmometry (mm)
-
23–123–28
-
-
tension
-
right: 15 mmHg
-
left: 15 mmHg
-
-
CT scan ([Fig. 21a, b])
-
on the left, evidence of four polylobulated, homogenously moderately contrast enhancing, intra- and extraconal lesions, the largest one measuring 18.5×26×18 mm
-
on the right, significantly smaller intramuscular lesions
-
-
-
Course
-
first, change in medication to cortisone-saving immunomodulated therapy initially with methotrexate, but because of intolerance shifted to mycophenolate mofetil; additional rheumatological care
-
because of no improvement, ENT-specific consultation and indication of balanced orbital decompression with intraorbital re-biopsy ([Fig. 22]) for verification of the diagnosis; transfer to the department of otolaryngology
-
-
Therapy
-
balanced medial and lateral orbital decompression on the left and biopsy of a mass (mostly intraconal location protruding in extraconal direction) via lateral orbitotomy
-
stepwise reduction of pharmacotherapy
-
-
Histopathological diagnosis
-
extranodal marginal zone lymphoma
-
-
Further course
-
postoperatively, slight visual improvement (0.5)
-
staging by means of whole-body CT scan and cMRT without hint of further manifestation of the lymphoma
-
case presentation to the tumor board and therapy by the departments of hemato-oncology and radiotherapy
-
local radiotherapy up to 30.6 Gy (ED 1.8 Gy) of the right and left orbits
-
cooperation with the department of hemato-oncology
-



#
7.2.2.2.3.2 Leukemia
-
Epidemiology
-
orbital manifestation in comparison to lymphomas clearly more rare
-
percentage of leukemias in malignant lymphoid orbital adnexa diseases: ~4% [365]
-
occurrence mostly during the first decade
-
-
Types
-
orbital manifestation possible in acute and chronic leukemias
-
most frequent subtype: acute myelic leukemia [466] [467] [468]
-
mostly secondary orbital involvement (e. g., as recurrence), orbital manifestation also a possible first symptom
-
bilateral occurrence possible
-
orbital involvement mostly a hint of an advanced disease stage
-
-
Therapy
-
chemotherapy and radiotherapy of the affected eye (better outcome compared to chemotherapy alone) [469]
-
#
7.2.2.2.4 Metastases [59] [470] [471]
-
Epidemiology
-
7–8% of all orbital neoplasms
-
in up to 25% of cases, evident before identification of the primary tumor
-
-
Differential diagnoses
-
clinically and morphologically, confusion with other diseases possible, for example, abscesses or parasitosis [471]
-
-
Location of the primary tumor
-
in adults mainly in the breast, lung, gastrointestinal tract, prostate, kidney, skin
-
in adults with involvement of the external eye muscles in the breast, skin (melanoma) [471]
-
in children mainly neuroblastoma, Ewing sarcoma
-
-
Therapy and management
-
identification of the primary tumor
-
depending on the tissue type
-
radiotherapy as significant therapeutic pillar (at least for symptom control)
Case report 5
-
-
History
-
62-year-old male patient
-
for 4 days, aqueous rhinorrhea, epiphora, increasing periorbital swelling and reddening, increasing pain in the left eye
-
no infection of the upper airways
-
-
Findings
-
reddening and swelling of the left upper and lower eyelid with extension to the cheek
-
exophthalmos
-
chemosis with conjunctival hyperemia
-
motility impairment of the bulb
-
reduced vision (0.6)
-
intraocular pressure: 14 mmHg on right, 20 mmHg on left
-
leukocytes and CRP slightly increased
-
CT scan (with contrast agent; [Fig. 23a, b])
-
lesion 12×11×10 mm, strongly contrast enhancing in the area of the inferior rectus muscle, slightly displacing the optic nerve
-
regularly ventilated paranasal sinuses
-
-
-
Course
-
because of the suspicion of intraorbital abscess, transnasal, transethmoid endoscopic orbitotomy with opening of the lesion releasing only limited turbid secretions; swab and biopsy performed
-
histology: neuroendocrine tumor
-
diagnosis after identification of the primary tumor, and staging (among others DOTA-TOC PET-CT): neuroendocrine tumor (G2) of the ileum with hepatic, orbital, and lymph node metastases
-

#
7.2.2.2.5 Secondary orbital tumors [59] [472]
-
Origin
-
paranasal sinuses
-
classification: grade I (infiltration of the orbital bone), grade II (infiltration of the periorbita), grade III (infiltration of the orbital soft tissue by transgression of the periorbita)
-
therapy: multimodal approach required (among others R0 resection, which is crucial for prognosis)
-
prognosis: with grade III, improved survival by orbital exenteration compared to organ preservation
-
-
intracranial
-
eyelid (e. g., basal cell carcinoma)
-
conjunctiva (e. g., melanoma)
-
#
#
#
7.2.3 Vascular anomalies
Vascular anomalies have a special position and can be classified into vascular tumors and malformations.
7.2.3.1 Vascular tumors
Vascular tumors have the following characteristics:
-
“True” neoplasms with endothelial proliferation and angiogenesis
-
Capacity for regression (at least partly) with increasing age
-
Differentiation of benign, locally aggressive (borderline), and malignant subtypes
7.2.2.2.5 Infantile hemangioma [2] [473] [474] [475]’

-
Synonym: capillary hemangioma (former term)
-
Epidemiology
-
most frequent vascular orbital tumor in childhood
-
manifestation typically in the first weeks of life (not at birth) with progression during the first 3–6 months
-
spontaneous regression typically from the 6th month to the 9th year of life (75–90% gone by the age of 7 years [476]), rarely only during puberty
-
in general harmless, but in cases of unfavorable location (e. g., orbit, oral cavity), possible functional impairment and permanent deformity (e. g., irreversible amblyopia, astigmatism)
-
-
Location and appearance ([Fig. 24a, b])
-
ranging from small and circumscribed to large and multifocal
-
most frequently extraconal, mostly ventral to the bulbus in the area of the eyelids; extraocular muscles and lacrimal gland also possibly affected
-
more rarely, intraconal, e. g., in the area of the orbital fat body [477]
-
in cases of retrobulbar location, risk of compression of the optic nerve (especially with rapid growth)
-
-
Clinical
-
raspberry-like appearance, especially with superficial location
-
-
Differential diagnosis
-
congenital hemangioma
-
much rarer, absolute rarity in the orbit
-
present already at birth and completely developed (no further growth after birth)
-
-
-
Therapy [478]
-
spontaneous remission in 85–90% of cases
-
treatment only in symptomatic hemangioma cases necessary; in cases of optic nerve compression, therapy urgently needed
-
propranolol 2–3 mg/kg body weight per day applied in 2–4 single doses for 6 months (therapy of first choice in cases of complex infantile hemangiomas; response rate of 98%)
-
meanwhile, systemic steroid application reserved for exceptional cases (e. g., airway obstruction, non-response to other therapeutic procedures)
-
cytostatics (vincristine) and interferon-alpha are obsolete
-
laser/cryotherapy: in small, plain, easily accessible hemangiomas (e. g., eyelid)
-
surgical resection: indicated rather rarely in the area of the orbit because of the risk of damaging surrounding structures
-
#
7.2.3.1.2 Hemangiopericytoma [479] [480]
-
Definition
-
tumor originating from the pericytes of the vascular wall or pluripotent perivascular cells
-
-
Epidemiology
-
only single reports of orbital occurrence [480]
-
-
Diagnostics [480]
-
no pathognomonic clinical or radiological characteristics
-
biopsy required
-
-
Pathology
-
histologically, the whole spectrum of benign to malignant characteristics ->classification into benign, borderline, and malignant
-
behavior rather unpredictable, with even seemingly benign lesions sometimes showing malignant behavior
-
metastasis in about 15% of cases
-
hemangiopericytoma and solitary fibrous tumors overlapping considerably in histology and immunohistochemistry, and according to some authors, possibly representing an independent entity [479]
-
-
Therapy
-
complete surgical resection
-
if needed, adjuvant radiotherapy
-
if needed, chemotherapy
-
follow-up
-
-
Prognosis
-
local recurrences in about 30% of cases
-
[Table 10] lists examples of other vascular tumors of the orbit. Most are absolute rarities in the orbit, and only single case reports are available [475]. Meanwhile, many authors classify tufted angioma as a borderline tumor and an independent entity together with Kaposi-form hemangioendothelioma – with a histopathological spectrum that has more or less aggressive characteristics, depending on the individual case [494] [495].
|
Status |
Vascular tumors |
|---|---|
|
Benign |
|
|
Locally aggressive/borderline |
|
|
Malignant |
* In the ISSVA classification, the tufted angioma is listed with the benign tumors; however, it must be considered as a borderline tumor forming an entity together with the Kaposi-form hemangioendothelioma.
#
#
7.2.3.2 Vascular malformations [2] [473] [474] [475] [495]
Vascular malformations have the following characteristics:
-
Mesenchymal vasculogenetic developmental disorders with low proliferative activity
-
Always present at birth but apparent only later (growth triggered, for example, by hormones or mechanical impact)
-
Progressive
-
No spontaneous remission
The classification of vascular malformations and therapeutic options is listed in [Table 11]. The percentages are general and not specific for the orbit [2] [475] ([Fig. 25a, b]).

|
Classification |
Therapy [474] |
|---|---|
|
Simple vascular malformations |
|
|
Slow-flow |
|
|
Sclerotherapy, laser therapy, resection |
|
Sclerotherapy (macrocystic), laser therapy, resection, mTOR inhibitors (sirolimus) [488] |
|
Laser therapy |
|
Fast-flow |
|
|
Embolization (transarterial, transvenous, direct), resection of the occluded nidus (=nucleus containing the shunt vessels) |
|
Arteriovenous fistula |
Embolization |
|
Combined vascular malformations (6%) |
|
|
7.2.3.2.1 Venous malformation [201] [412]
-
Synonym
-
former ‘cavernous hemangioma’, and according to the current ISSVA classification of vascular anomalies [494], to be called ‘slow-flow venous malformation’ (however, the former term is still widely used)
-
-
Epidemiology
-
one of the most frequent benign masses of the orbit in adults (5–7% of all orbital lesions)
-
typical age of manifestation: 30–50 years
-
women more frequently affected
-
-
Clinical
-
often incidental finding
-
slow growth (exophthalmos developing over years)
-
increased growth during pregnancy
-
typical location: intraconal
-
-
Therapy
-
“watch-and-wait” in smaller/asymptomatic findings
-
sclerotherapy
-
complete surgical resection, e. g., via lateral orbitotomy
-
-
Differential diagnosis
-
‘orbital varicosities’
-
the term ‘orbital varicosities’ is no longer used, and they are classified as venous malformations
-
pathogenesis: congenital weakness of the venous walls of the valve-less orbital vein ->massive dilation
-
diagnostics: exophthalmos during Valsalva maneuver
-
Clinical: in cases of bleeding, rapid and painful increase in exophthalmos
-
therapy: in general, not required
-
-
#
7.2.3.2.2 Lymphatic malformation [412] [473]
-
Synonym (no longer used)
-
lymphangioma
-
-
Epidemiology
-
occurring mainly in childhood and adolescence
-
-
Clinical
-
periorbital swelling
-
slowly progressive, indolent exophthalmos
-
motility disorders
-
bulbus displacement
-
complication: acute exacerbation because of bleeding or infection possible (significant increase within hours or days also possible)
-
no capsula, diffusely infiltrating, compartment borders not respected
-
filled with lymphatic fluid or blood
-
-
Therapy
-
complete extirpation usually not possible
-
“watch-and-wait”
-
in cases of risk of vision loss, volume-reducing measures such as partial resection, injection of sclerotizing substances, mTOR inhibitors
-
#
7.2.3.2.3 Arteriovenous malformation (AVM) [473] [475] [495] [496]

-
Location
-
mainly in the head and neck, including brain ([Fig. 26])
-
extremities
-
more rarely, inner organs (e. g., liver, lung)
-
-
Characteristics
-
clinically quite diverse and challenging
-
unexceptionally life-long progressive growth (destructive)
-
reacts to stimuli such as hormonal changes, trauma (e. g., possible increased proliferation after incomplete invasive therapy) [474]
-
-
Clinical classification (Schobinger classification, shortened) [497]
-
stage I: clinically inactive
-
stage II: expansion, pulsation
-
stage III: destructive AVM with ulcerations, necrosis, bleeding, pain, infection
-
stage IV: decompensated AVM with heart failure
-
-
Therapy
-
often quite difficult and risky
-
“watch-and-wait” in cases of asymptomatic AVM
-
possibly complete closure by embolization (several sessions) with subsequent resection (if the location allows) to avoid recurrence
-
mTOR inhibitors not effective
-
-
Prognosis
-
high tendency to recurrence in cases of incomplete therapy
Case report 6
-
-
History
-
emergency presentation of a 9-month-old boy with acute right-sided exophthalmos
-
no relevant previous diseases
-
-
Diagnostics
-
inspection: right-sided exophthalmos, increased vascular injection, incomplete eyelid closure ([Fig. 27])
-
ultrasound at first presentation: suspicion of retrobulbar, acutely bleeding, cavernous venous malformation, MRI for further clarification recommended
-
MRI ([Fig. 28]): retrobulbar bleeding with suspected ruptured, partly thrombosed aneurysma spurium with additional venous malformation portions, DD AVM
-
angiography: high-flow AVM in the right-sided orbit, mainly fed from the right ophthalmic artery, dominant venous outflow via dilated venous aneurysms (type II according to the WHO classification), venous drainage via the cavernous sinus
-
-
Course
-
progressive exophthalmos, right-sided
-
increasing defense of the child
-
severe right-sided loss of vision
-
-
Therapy
-
embolization of the AVM with onyx18 (interventional radiology) ->complete closure of the inflow via the ophthalmic artery and another feeder from the maxillary artery ->AVM considered operable
-
because of progressive exophthalmos and onset of corneal ulceration of the right eye, interdisciplinary case discussion and indication for surgery
-
bulbus enucleation, resection of the intraconal AVM after embolization, implantation of a Guthoff implant (16 mm) with tenon and conjunctiva plasty (interdisciplinary by ophthalmologists and otolaryngologists; [Fig. 29])
-
later epithetic treatment
-
-
Histology
-
AVM with condition after embolization
-
no hint of malignancy
-


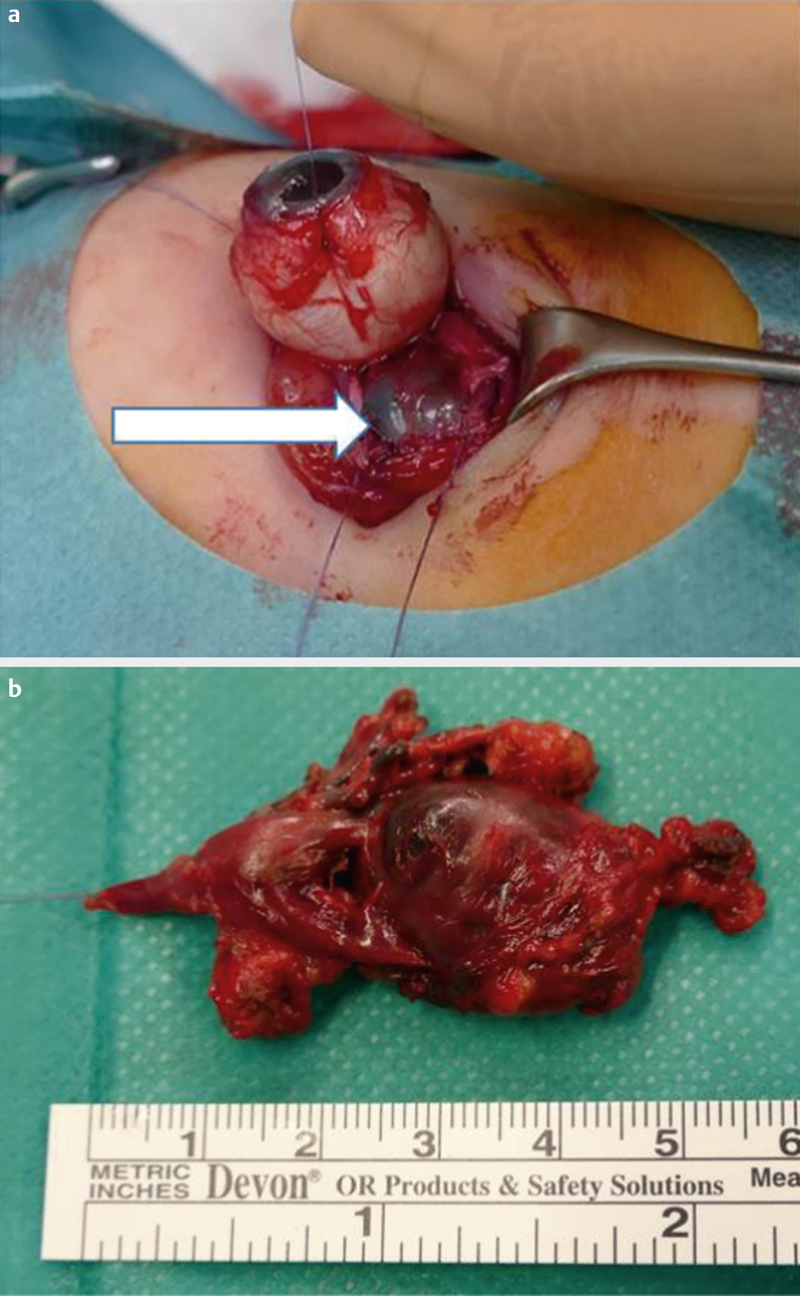
#
#
#
#
7.3. Conclusion
Neoplasms of the orbit are rare but diverse. Based on symptoms alone, they cannot be differentiated from structural or inflammatory lesions, and differentiated ophthalmological, radiological, and paraclinical diagnostics are important. In most cases, however, histopathological examination of the excisional biopsy is decisive. Exceptions are tumors of epithelial origin of the lacrimal gland, in which incision biopsies may significantly worsen the overall prognosis. The classification of orbital tumors consists of neoplasms of epithelial and non-epithelial origin as well as neoplasms of the lymphatic tissue, whereas the lacrimal gland is the only organ of the orbit with epithelial structures. Vascular malformations are a special case because they have a highly variable clinical behavior. In addition to watch-and-wait in asymptomatic benign processes, surgical resection of symptomatic benign neoplasms is mainly considered. However, especially in the area of the orbit, the expected benefit must be weighed against the possible functional deficits. The access pathways mainly depend on the location of the lesions. Radiotherapy as well as systemic therapies with chemotherapeutics and biologicals are important therapeutic pillars and in recent decades have reduced morbidity with orbital tumors. Nonetheless, malignant orbital tumors are mostly associated with a poor prognosis.
Lymphoproliferative lesions of the orbit are indolent so that patients often have a good quality of life after remission. However, mortality rates can be quite high, for example in cases of NK/T cell lymphomas. A better understanding of the molecular genetic correlations will certainly lead to more precise application of targeted therapeutics and thus to a better prognosis with orbital lymphomas. An essential condition, however, is conducting biopsies that are actually representative.
Bone tumors of the orbit encompass highly variable entities, and fibro-osseous lesions have fascinating pathogenetic and pathological properties. Imaging allows differentiation among the possible diagnoses, but biopsy is often necessary for confirmation. These evaluations are reserved mainly for referral centers. Treatment decisions should always be made with consideration of the systemic effects of the disease and new approaches with targeted therapies.
Because of their rarity and diversity, tumors of the lacrimal gland are challenging not only for pathologists but also for clinicians. History and symptom assessment are crucial for the diagnosis but can vary enormously, and even imaging is rarely pathognomonic. Furthermore, controversies exist with regard to the most appropriate therapies, especially in the context of malignant epithelial tumors that generally have a poorer prognosis compared to malignancies of the salivary glands.
Primary and secondary neurogenic tumors are often located in the orbit, presumably because of the high density of nerves in this region. Diagnostics and therapy represent a challenge given that the etiology, pathogenesis, and natural course of the diseases are still not clear.
Tumors of the orbital fatty and connective tissues as well as the striated muscles are mostly heterogeneous neoplasms that are nonetheless similar with regard to clinical symptoms, histogenetics, molecular biology, and prognosis.
Metastases of the orbit are another heterogeneous group of neoplasms with a high variation in presentation, depending on the disease extent and biology of the primary tumor. Most metastases occur in patients with an unknown primary tumor and become apparent because of displaced bulbus, strabismus, and pain. In cases of suspicion, imaging and biopsy are necessary to plan multidisciplinary treatment aimed at preserving vision, reducing pain, and improving quality of life. In some cases, a curative approach is possible.
Vascular anomalies are subdivided into vascular tumors and vascular malformations. Although the former are “true” neoplasms with endothelial proliferation and angiogenesis, malformations are vasculogenetic developmental disorders. They have a low proliferative activity and are already present at birth but become apparent only later and do not show spontaneous remission. The heterogenous clinical presentation of vascular tumors requires clarification of various differential diagnoses. Therapeutic alternatives span a large spectrum that includes pharmacotherapy, laser, and classic surgical interventions so that most patients might benefit from one of these options. Dural AV fistulas and carotid cavernous fistulas may have variable orbital manifestations. Despite differences in the lesions, the same examination procedures are applied. Characterization of a lesion and therapy planning require conventional catheter angiography. In benign cases, the therapeutic strategy is conservative, but in rapidly progressing and severe cases, neuroendovascular management is preferred.
#
#
#
Interessenkonflikt
Die Autorinnen/Autoren geben an, dass kein Interessenkonflikt besteht.
Acknowledgements
The authors thank Prof. H.-J. Welkoborsky, Dr. C. Adderson-Kisser, Dr. I. Seiwerth, A. Gey, Prof. W. Wohlgemuth, A. Troch, and Prof. S. K. Plontke for their support in the preparation of this manuscript.
-
Literatur
- 1 Poloschek CM, Lagreze WA, Ridder GJ. et al. [Clinical and neuroradiological diagnostics of orbital tumors]. Ophthalmologe 2011; 108: 510-518
- 2 Plontke SK, Glien A, Kisser U. et al. [Diseases and Surgery of the Orbit]. Laryngorhinootologie 2020; 99: 896-917
- 3 AWMF. Rekonstruktion von Orbitadefekten (S2e-Leitlinie), Registernummer: 007/099. In: Arbeitsgemeinschaft medizinisch wissenschaftlicher Fachgesellschaften (AWMF); 2013
- 4 Fay A, Dolman PJ. Laboratory Serologic Investigations.In Fay A, Dolman PJ , Hrsg. Diseases and Disorders of the Orbit and Ocular Adnexa E-book. Elsevier; 2017: 57–77
- 5 Bonavolonta G, Tranfa F, de Conciliis C. et al. Dermoid cysts: 16-year survey. Ophthalmic Plast Reconstr Surg 1995; 11: 187-192
- 6 Jordan DR, Spitellie P, Brownstein S. et al. Orbital cholesterol granuloma and cholesteatoma: significance of differentiating the two. Ophthalmic Plast Reconstr Surg 2007; 23: 415-417
- 7 Sherman RP, Rootman J, Lapointe JS. Orbital dermoids: clinical presentation and management. Br J Ophthalmol 1984; 68: 642-652
- 8 Shields JA, Kaden IH, Eagle RC. et al. Orbital dermoid cysts: clinicopathologic correlations, classification, and management. The 1997 Josephine E. Schueler Lecture. Ophthalmic Plast Reconstr Surg 1997; 13: 265-276
- 9 Delfini R, Missori P, Iannetti G. et al. Mucoceles of the paranasal sinuses with intracranial and intraorbital extension: report of 28 cases. Neurosurgery 1993; 32: 901-906 discussion 906
- 10 Heichel J, Struck HG, Hammer T. et al. [Pediatric acute dacryocystitis due to frontoethmoidal mucocele]. HNO 2019; 67: 458-462
- 11 Nicollas R, Facon F, Sudre-Levillain I. et al. Pediatric paranasal sinus mucoceles: etiologic factors, management and outcome. Int J Pediatr Otorhinolaryngol 2006; 70: 905-908
- 12 Holtmann C, Brachert M, Spaniol K. et al. [Dacryolith in a lacrimal duct]. Ophthalmologe 2016; 113: 244-247
- 13 Tanaboonyawat S, Idowu OO, Copperman TS. et al. Dacryops - A review. Orbit 2020; 39: 128-134
- 14 Junemann A, Holbach LM. [Epithelial giant inclusion cyst 50 years after enucleation without orbital implant]. Klin Monbl Augenheilkd 1998; 212: 127-128
- 15 Kalantzis GK, Verity DH, Rose GE. Periocular implantation cysts: a late complication of ophthalmic surgery. Eye (Lond) 2014; 28: 1004-1007
- 16 Gupta R, Seith A, Guglani B. et al. Congenital cystic eye: features on MRI. Br J Radiol 2007; 80: e137-140
- 17 Guthoff R, Klein R, Lieb WE. Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 2004; 242: 268-271
- 18 Kuchle HJ, Normann J, Lubbering I. [Congenital cystic eye]. Klin Monbl Augenheilkd 1986; 188: 239-241
- 19 Schittkowski MP, Guthoff RF. Injectable self inflating hydrogel pellet expanders for the treatment of orbital volume deficiency in congenital microphthalmos: preliminary results with a new therapeutic approach. Br J Ophthalmol 2006; 90: 1173-1177
- 20 Schittkowski MP, Guthoff RF. Systemic and ophthalmological anomalies in congenital anophthalmic or microphthalmic patients. Br J Ophthalmol 2010; 94: 487-493
- 21 Badtke G, Tost M. Anophthalmus congenitus.In Velhagen K , Hrsg. Der Augenarzt, Band XI. Leipzig: VEB Georg Thieme; 1986: 328–344
- 22 Badtke G, Tost M. Mikrophthalmus congenitus.In Velhagen K , Hrsg. Der Augenarzt, Band XI. Leipzig: VEB Georg Thieme; 1986: 312–328
- 23 Chaudhry IA, Arat YO, Shamsi FA. et al. Congenital microphthalmos with orbital cysts: distinct diagnostic features and management. Ophthalmic Plast Reconstr Surg 2004; 20: 452-457
- 24 Oguz H, Ozturk A, San I. Congenital nasolacrimal duct occlusion with clinical anophthalmos: a possible new association. Ophthalmic Genet 2003; 24: 181-185
- 25 Pejic M, Luecke K, Meoded A. et al. Pediatric Cephaloceles: A Multimodality Review. Appl Radiol 2020; 49: 26-32
- 26 Terry A, Patrinely JR, Anderson RL. et al. Orbital meningoencephalocele manifesting as a conjunctival mass. Am J Ophthalmol 1993; 115: 46-49
- 27 Vashisht S, Ghai S, Hatimota P. et al. Cystic lesions of the orbit : A CT spectrum. Indian Journal of Radiology and Imaging 2003; 13: 139
- 28 Modi P, Shah NA, Bhalodia JN. et al. ORBITAL TUMORS IN CHILDREN: A DESCRIPTIVE STUDY AT TERTIARY CARE CENTRE. The Journal of medical research 2013; 3: 362-366
- 29 Green WR, Zimmerman LE. Ectopic lacrimal gland tissue. Report of eight cases with orbital involvement. Arch Ophthalmol 1967; 78: 318-327
- 30 Scheiner AJ, Frayer WC, Rorke LB et al. Ectopic brain tissue in the orbit. Eye (Lond) 1999; 13: ( Pt 2) 251–254
- 31 Damato PJ, Damato FJ. Neonatal Orbital Teratoma. Br J Ophthalmol 1962; 46: 685-691
- 32 Herman TE, Vachharajani A, Siegel MJ. Massive congenital orbital teratoma. J Perinatol 2009; 29: 396-397
- 33 Sesenna E, Ferri A, Thai E. et al. Huge orbital teratoma with intracranial extension: a case report. J Pediatr Surg 2010; 45: e27-31
- 34 Duke-Elder S. Anomalies of the Bony Orbit. In: Duke-Elder S, Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964: 942-949
- 35 Exner S, Bogusch G, Sokiranski R. Cribra orbitalia visualized in computed tomography. Ann Anat 2004; 186: 169-172
- 36 Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol 2004; 88: 833-834
- 37 Duke-Elder S. Deformities of the Face.In Duke-Elder S , Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964; 1007–1035
- 38 Kawamoto HK. The kaleidoscopic world of rare craniofacial clefts: order out of chaos (Tessier classification). Clin Plast Surg 1976; 3: 529-572
- 39 Fearon JA. Rare craniofacial clefts: a surgical classification. J Craniofac Surg 2008; 19: 110-112
- 40 Tessier P. Anatomical classification facial, cranio-facial and latero-facial clefts. J Maxillofac Surg 1976; 4: 69-92
- 41 Heike CL, Hing AV, Aspinall CA. et al. Clinical care in craniofacial microsomia: a review of current management recommendations and opportunities to advance research. Am J Med Genet C Semin Med Genet 2013; 163C: 271-282
- 42 Dupont S, Catala M, Hasboun D. et al. Progressive facial hemiatrophy and epilepsy: a common underlying dysgenetic mechanism. Neurology 1997; 48: 1013-1018
- 43 Fea AM, Aragno V, Briamonte C. et al. Parry Romberg syndrome with a wide range of ocular manifestations: a case report. BMC Ophthalmol 2015; 15: 119
- 44 Duke-Elder S. Deformities of the Skull.In Duke-Elder S , Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964: 1035–1072
- 45 Freudlsperger C, Castrillón-Oberndorfer G, Hoffmann J. et al. Isolierte, nichtsyndromale Kraniosynostosen. Der MKG-Chirurg 2013; 6: 301-313
- 46 Governale LS. Craniosynostosis. Pediatr Neurol 2015; 53: 394-401
- 47 Kotrikova B, Muhling J. [Surgery of craniofacial deformities]. Klin Monbl Augenheilkd 2004; 221: 970-977
- 48 Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr 2017; 29: 622-628
- 49 Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet 1999; 36: 177-182
- 50 Dubourg C, Bendavid C, Pasquier L. et al. Holoprosencephaly. Orphanet J Rare Dis 2007; 2: 8
- 51 Lustig LR, Holliday MJ, McCarthy EF. et al. Fibrous dysplasia involving the skull base and temporal bone. Arch Otolaryngol Head Neck Surg 2001; 127: 1239-1247
- 52 Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis 2009; 4: 5
- 53 Lannon DA, Earley MJ. Cherubism and its charlatans. Br J Plast Surg 2001; 54: 708-711
- 54 Grass S, Welkoborsky HJ, Mobius H. et al. [Orbital complications]. HNO 2018; 66: 800-811
- 55 Lehnerdt G, Peraud A, Berghaus A. et al. [Orbital and intracranial complications of acute sinusitis. Diagnostics and therapy in children and adolescents]. HNO 2011; 59: 75-86 quiz 87-78
- 56 Stammberger H. [Complications of inflammatory paranasal sinus diseases including iatrogenically-induced complications]. Eur Arch Otorhinolaryngol Suppl 1993; 1: 61-102
- 57 Welkoborsky HJ, Grass S, Deichmuller C. et al. Orbital complications in children: differential diagnosis of a challenging disease. Eur Arch Otorhinolaryngol 2015; 272: 1157-1163
- 58 Hamed-Azzam S, AlHashash I, Briscoe D. et al. Rare Orbital Infections ~ State of the Art ~ Part II. J Ophthalmic Vis Res 2018; 13: 183-190
- 59 Rootman J. Diseases of the orbit.Second edition Aufl. Lippincott Williams & Wilkins; Philadelphia: 2003
- 60 Madge SN, Prabhakaran VC, Shome D. et al. Orbital tuberculosis: a review of the literature. Orbit 2008; 27: 267-277
- 61 Fay A, Dolman PJ. Diseases and Disorders of the Orbit and Ocular Adnexa Expert Consult. In Fay A, Dolman PJ (eds). Elsevier; 2017
- 62 Heichel J, Griebsch M, Siebolts U. et al. [Bilateral periocular necrotizing fasciitis with unilateral orbital involvement]. Ophthalmologe 2020; 117: 1121-1125
- 63 Huber L, Budjan J, Rotter N. et al. [Necrotizing fasciitis in the head and neck region-three case reports and a review of the literature]. HNO 2020; 68: 935-943
- 64 Amrith S, Hosdurga Pai V, Ling WW. Periorbital necrotizing fasciitis -- a review. Acta Ophthalmol 2013; 91: 596-603
- 65 Wong CH, Chang HC, Pasupathy S. et al. Necrotizing fasciitis: clinical presentation, microbiology, and determinants of mortality. J Bone Joint Surg Am 2003; 85: 1454-1460
- 66 Lazzeri D, Lazzeri S, Figus M. et al. Periorbital necrotising fasciitis. Br J Ophthalmol 2010; 94: 1577-1585
- 67 Wladis EJ, Levin F, Shinder R. Clinical Parameters and Outcomes in Periorbital Necrotizing Fasciitis. Ophthalmic Plast Reconstr Surg 2015; 31: 467-469
- 68 Federspil P. German Society of Oto-Rhino-Laryngology H, Neck S. [Guidelines: Antibiotic treatment of infections of the head and neck: consensus report on behalf of the presidency of the German Society of Oto-Rhino-Laryngology, Head and Neck Surgery edited by P. Federspil, Homburg/Saar]. . HNO 2009; 57: 377-394
- 69 Barrani M, Massei F, Scaglione M. et al. Unusual onset of a case of chronic recurrent multifocal osteomyelitis. Pediatr Rheumatol Online J 2015; 13: 60
- 70 Dagan O, Barak Y, Metzker A. Pyoderma gangrenosum and sterile multifocal osteomyelitis preceding the appearance of Takayasu arteritis. Pediatr Dermatol 1995; 12: 39-42
- 71 Huber AM, Lam PY, Duffy CM. et al. Chronic recurrent multifocal osteomyelitis: clinical outcomes after more than five years of follow-up. J Pediatr 2002; 141: 198-203
- 72 Hu AC, Ng WKY. Orbital Osteomyelitis in the Pediatric Patient. J Craniofac Surg 2021; 32: 206-209
- 73 Cornely OA, Alastruey-Izquierdo A, Arenz D. et al. Global guideline for the diagnosis and management of mucormycosis: an initiative of the European Confederation of Medical Mycology in cooperation with the Mycoses Study Group Education and Research Consortium. Lancet Infect Dis 2019; 19: e405-e421
- 74 Bakhshaee M, Bojdi A, Allahyari A. et al. Acute invasive fungal rhinosinusitis: our experience with 18 cases. Eur Arch Otorhinolaryngol 2016; 273: 4281-4287
- 75 van Burik JA, Hare RS, Solomon HF. et al. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42: e61-65
- 76 Songu M, Unlu HH, Gunhan K. et al. Orbital exenteration: A dilemma in mucormycosis presented with orbital apex syndrome. Am J Rhinol 2008; 22: 98-103
- 77 Nicolai P, Lombardi D, Tomenzoli D. et al. Fungus ball of the paranasal sinuses: experience in 160 patients treated with endoscopic surgery. Laryngoscope 2009; 119: 2275-2279
- 78 Johnson TE, Casiano RR, Kronish JW. et al. Sino-orbital aspergillosis in acquired immunodeficiency syndrome. Arch Ophthalmol 1999; 117: 57-64
- 79 Levin LA, Avery R, Shore JW. et al. The spectrum of orbital aspergillosis: a clinicopathological review. Surv Ophthalmol 1996; 41: 142-154
- 80 Siddiqui AA, Shah AA, Bashir SH. Craniocerebral aspergillosis of sinonasal origin in immunocompetent patients: clinical spectrum and outcome in 25 cases. Neurosurgery 2004; 55: 602-611 discussion 611-603
- 81 Nielsen EW, Weisman RA, Savino PJ. et al. Aspergillosis of the sphenoid sinus presenting as orbital pseudotumor. Otolaryngol Head Neck Surg 1983; 91: 699-703
- 82 Sivak-Callcott JA, Livesley N, Nugent RA. et al. Localised invasive sino-orbital aspergillosis: characteristic features. Br J Ophthalmol 2004; 88: 681-687
- 83 Lamoth F, Calandra T. Early diagnosis of invasive mould infections and disease. J Antimicrob Chemother 2017; 72: i19-i28
- 84 McCarthy M, Rosengart A, Schuetz AN. et al. Mold infections of the central nervous system. N Engl J Med 2014; 371: 150-160
- 85 Green WR, Font RL, Zimmerman LE. Asperillosis of the orbit. Report of ten cases and review of the literature. Arch Ophthalmol 1969; 82: 302-313
- 86 Denning DW. Therapeutic outcome in invasive aspergillosis. Clin Infect Dis 1996; 23: 608-615
- 87 Robert Koch-Institut. Infektionsepidemiologisches Jahrbuch meldepflichtiger Krankheiten für 2019 Berlin: Robert Koch-Institut; 2020
- 88 Gomez Morales A, Croxatto JO, Crovetto L. et al. Hydatid cysts of the orbit. A review of 35 cases. Ophthalmology 1988; 95: 1027-1032
- 89 Xiao A, Xueyi C. Hydatid cysts of the orbit in Xinjiang: a review of 18 cases. Orbit 1999; 18: 151-155
- 90 Kars Z, Kansu T, Ozcan OE. et al. Orbital echinococcosis. Report of two cases studied by computerized tomography. J Clin Neuroophthalmol 1982; 2: 197-199
- 91 Chtira K, Benantar L, Aitlhaj H. et al. The surgery of intra-orbital hydatid cyst: a case report and literature review. Pan Afr Med J 2019; 33: 167
- 92 Senyuz OF, Yesildag E, Celayir S. Albendazole therapy in the treatment of hydatid liver disease. Surg Today 2001; 31: 487-491
- 93 Pushker N, Bajaj MS, Balasubramanya R. Disseminated cysticercosis involving orbit, brain and subcutaneous tissue. J Infect 2005; 51: e245-248
- 94 Walrath JD, Lalin SC, Leib ML. Cysticercosis isolated to the orbit. Ophthalmic Plast Reconstr Surg 2003; 19: 243-244
- 95 Sotelo J, Escobedo F, Rodriguez-Carbajal J. et al. Therapy of parenchymal brain cysticercosis with praziquantel. N Engl J Med 1984; 310: 1001-1007
- 96 Nentwich MM, Pleyer U, Schaller UC. et al. [International ophthalmology and travel medicine]. Ophthalmologe 2016; 113: 83-94
- 97 Mombaerts I, Rose GE, Garrity JA. Orbital inflammation: Biopsy first. Surv Ophthalmol 2016; 61: 664-669
- 98 Ben Simon GJ, Yoon MK, Atul J. et al. Clinical manifestations of orbital mass lesions at the Jules Stein Eye Institute, 1999-2003. Ophthalmic Surg Lasers Imaging 2006; 37: 25-32
- 99 Nölle B, Both M. Idiopathische Orbitaentzündung.In Pleyer U , Hrsg. Entzündliche Augenerkrankungen. Berlin, Heidelberg: Springer- Verlag; 2016: 48–57
- 100 Swamy BN, McCluskey P, Nemet A. et al. Idiopathic orbital inflammatory syndrome: clinical features and treatment outcomes. Br J Ophthalmol 2007; 91: 1667-1670
- 101 Fujii H, Fujisada H, Kondo T. et al. Orbital pseudotumor: histopathological classification and treatment. Ophthalmologica 1985; 190: 230-242
- 102 Mombaerts I, Goldschmeding R, Schlingemann RO. et al. What is orbital pseudotumor?. Surv Ophthalmol 1996; 41: 66-78
- 103 Rootman J, Nugent R. The classification and management of acute orbital pseudotumors. Ophthalmology 1982; 89: 1040-1048
- 104 Rootman J, McCarthy M, White V. et al. Idiopathic sclerosing inflammation of the orbit. A distinct clinicopathologic entity. Ophthalmology 1994; 101: 570-584
- 105 Iaconetta G, Stella L, Esposito M. et al. Tolosa-Hunt syndrome extending in the cerebello-pontine angle. Cephalalgia 2005; 25: 746-750
- 106 Speth MM, Metternich FU, Mina R. et al. Tolosa-Hunt-Syndrom, das sich als migrierende, auf Kortikosteroid reagierende CN3- und CN6-Lähmung bei verschatteten Keilbeinhöhlen darstellt. forum hno: omnimed Verlag 05/2020;
- 107 Halabi T, Sawaya R. Successful Treatment of Tolosa-Hunt Syndrome after a Single Infusion of Infliximab. J Clin Neurol 2018; 14: 126-127
- 108 Mormont E, Laloux P, Vauthier J. et al. Radiotherapy in a case of Tolosa-Hunt syndrome. Cephalalgia 2000; 20: 931-933
- 109 Perez CA, Evangelista M. Evaluation and Management of Tolosa-Hunt Syndrome in Children: A Clinical Update. Pediatr Neurol 2016; 62: 18-26
- 110 Zhang X, Zhang W, Liu R. et al. Factors that influence Tolosa-Hunt syndrome and the short-term response to steroid pulse treatment. J Neurol Sci 2014; 341: 13-16
- 111 Espinoza GM, Liu JL. Orbital Vasculitides-Differential Diagnosis. Curr Rheumatol Rep 2019; 21: 54
- 112 Tiegs-Heiden CA, Eckel LJ, Hunt CH. et al. Immunoglobulin G4-related disease of the orbit: imaging features in 27 patients. AJNR Am J Neuroradiol 2014; 35: 1393-1397
- 113 Erdei A, Steiber Z, Molnar C. et al. Exophthalmos in a young woman with no graves' disease - a case report of IgG4-related orbitopathy. BMC Ophthalmol 2018; 18: 5
- 114 Jurkov M, Olze H, Klauschen F. et al. [IgG4-Related Orbitopathy as an Important Differential Diagnosis of Advanced Silent Sinus Syndrome. German version]. HNO 2020; 68: 864-868
- 115 Mulholland GB, Jeffery CC, Satija P. et al. Immunoglobulin G4-related diseases in the head and neck: a systematic review. J Otolaryngol Head Neck Surg 2015; 44: 24
- 116 Detiger SE, Karim AF, Verdijk RM. et al. The treatment outcomes in IgG4-related orbital disease: a systematic review of the literature. Acta Ophthalmol 2019; 97: 451-459
- 117 Plaza JA, Garrity JA, Dogan A. et al. Orbital inflammation with IgG4-positive plasma cells: manifestation of IgG4 systemic disease. Arch Ophthalmol 2011; 129: 421-428
- 118 Perumal B, Black EH, Levin F. et al. Non-infectious orbital vasculitides. Eye (Lond) 2012; 26: 630-639
- 119 Sfiniadaki E, Tsiara I, Theodossiadis P. et al. Ocular Manifestations of Granulomatosis with Polyangiitis: A Review of the Literature. Ophthalmol Ther 2019; 8: 227-234
- 120 Mukhtyar C, Flossmann O, Hellmich B. et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis 2008; 67: 1004-1010
- 121 Holle JU, Voigt C, Both M. et al. Orbital masses in granulomatosis with polyangiitis are associated with a refractory course and a high burden of local damage. Rheumatology (Oxford) 2013; 52: 875-882
- 122 Perry SR, Rootman J, White VA. The clinical and pathologic constellation of Wegener granulomatosis of the orbit. Ophthalmology 1997; 104: 683-694
- 123 Chang SY, Keogh KA, Lewis JE. et al. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener's): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol 2013; 44: 2432-2437
- 124 Martinez Del Pero M, Chaudhry A, Jones RB. et al. B-cell depletion with rituximab for refractory head and neck Wegener's granulomatosis: a cohort study. Clin Otolaryngol 2009; 34: 328-335
- 125 Takanashi T, Uchida S, Arita M. et al. Orbital inflammatory pseudotumor and ischemic vasculitis in Churg-Strauss syndrome: report of two cases and review of the literature. Ophthalmology 2001; 108: 1129-1133
- 126 Cottin V, Cordier JF. Churg-Strauss syndrome. Allergy 1999; 54: 535-551
- 127 Masi AT, Hunder GG, Lie JT. et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990; 33: 1094-1100
- 128 Vaglio A, Moosig F, Zwerina J. Churg-Strauss syndrome: update on pathophysiology and treatment. Curr Opin Rheumatol 2012; 24: 24-30
- 129 Guillevin L, Lhote F, Gayraud M. et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996; 75: 17-28
- 130 Nagel MA, White T, Khmeleva N. et al. Analysis of Varicella-Zoster Virus in Temporal Arteries Biopsy Positive and Negative for Giant Cell Arteritis. JAMA Neurol 2015; 72: 1281-1287
- 131 Borruat FX, Bogousslavsky J, Uffer S. et al. Orbital infarction syndrome. Ophthalmology 1993; 100: 562-568
- 132 Stone JH, Tuckwell K, Dimonaco S. et al. Trial of Tocilizumab in Giant-Cell Arteritis. N Engl J Med 2017; 377: 317-328
- 133 Seyahi E. Takayasu arteritis: an update. Curr Opin Rheumatol 2017; 29: 51-56
- 134 Tugal-Tutkun I. Systemic vasculitis and the eye. Curr Opin Rheumatol 2017; 29: 24-32
- 135 Misra DP, Wakhlu A, Agarwal V. et al. Recent advances in the management of Takayasu arteritis. Int J Rheum Dis 2019; 22 (Suppl. 01) 60-68
- 136 Iliescu DA, Timaru CM, Batras M. et al. Cogan's Syndrome. Rom J Ophthalmol 2015; 59: 6-13
- 137 Tayer-Shifman OE, Ilan O, Tovi H. et al. Cogan's syndrome--clinical guidelines and novel therapeutic approaches. Clin Rev Allergy Immunol 2014; 47: 65-72
- 138 Grasland A, Pouchot J, Hachulla E. et al. Typical and atypical Cogan's syndrome: 32 cases and review of the literature. Rheumatology (Oxford) 2004; 43: 1007-1015
- 139 Gluth MB, Baratz KH, Matteson EL. et al. Cogan syndrome: a retrospective review of 60 patients throughout a half century. Mayo Clin Proc 2006; 81: 483-488
- 140 Padoan R, Cazzador D, Pendolino AL. et al. Cogan's syndrome: new therapeutic approaches in the biological era. Expert Opin Biol Ther 2019; 19: 781-788
- 141 Kokturk A. Clinical and Pathological Manifestations with Differential Diagnosis in Behcet's Disease. Patholog Res Int 2012; 2012: 690390
- 142 Hammami S, Yahia SB, Mahjoub S. et al. Orbital inflammation associated with Behcet's disease. Clin Exp Ophthalmol 2006; 34: 188-190
- 143 Urruticoechea-Arana A, Cobo-Ibanez T, Villaverde-Garcia V. et al. Efficacy and safety of biological therapy compared to synthetic immunomodulatory drugs or placebo in the treatment of Behcet's disease associated uveitis: a systematic review. Rheumatol Int 2019; 39: 47-58
- 144 Bettiol A, Silvestri E, Di Scala G. et al. The right place of interleukin-1 inhibitors in the treatment of Behcet's syndrome: a systematic review. Rheumatol Int 2019; 39: 971-990
- 145 Hedrich CM, Schnabel A, Hospach T. Kawasaki Disease. Front Pediatr 2018; 6: 198
- 146 Demirsoy S, Gucuyener K, Olgunturk R. et al. A case of Kawasaki syndrome associated with preseptal cellulitis in orbita. Turk J Pediatr 1988; 30: 55-59
- 147 Felz MW, Patni A, Brooks SE. et al. Periorbital vasculitis complicating Kawasaki syndrome in an infant. Pediatrics 1998; 101: E9
- 148 Sheard RM, Pandey KR, Barnes ND. et al. Kawasaki disease presenting as orbital cellulitis. J Pediatr Ophthalmol Strabismus 2000; 37: 123-125
- 149 Son MB, Gauvreau K, Burns JC. et al. Infliximab for intravenous immunoglobulin resistance in Kawasaki disease: a retrospective study. J Pediatr 2011; 158: 644-649 e641
- 150 Read RW. Clinical mini-review: systemic lupus erythematosus and the eye. Ocul Immunol Inflamm 2004; 12: 87-99
- 151 Arthurs BP, Khalil MK, Chagnon F. et al. Orbital infarction and melting in a patient with systemic lupus erythematosus. Ophthalmology 1999; 106: 2387-2390
- 152 Nabili S, McCarey DW, Browne B. et al. A case of orbital myositis associated with rheumatoid arthritis. Ann Rheum Dis 2002; 61: 938-939
- 153 Sanchez IJ, Finger DR, Bradshaw DJ. Orbital inflammatory syndrome and systemic lupus erythematosus. J Clin Rheumatol 1995; 1: 295-298
- 154 Kokotis P, Theodossiadis P, Bouros C. et al. Bilateral ocular myositis as a late complication of dermatomyositis. J Rheumatol 2005; 32: 379-381
- 155 Lokdarshi G, Pushker N, Bajaj MS. Sclerosing Lesions of the Orbit: A Review. Middle East Afr J Ophthalmol 2015; 22: 447-451
- 156 Petrarolha SM, Rodrigues BS, Filho FD. et al. Unilateral Eyelid Edema as Initial Sign of Orbital Sarcoidosis. Case Rep Ophthalmol Med 2016; 2016: 6912927
- 157 Rosenbaum JT, Choi D, Wilson DJ. et al. Parallel Gene Expression Changes in Sarcoidosis Involving the Lacrimal Gland, Orbital Tissue, or Blood. JAMA Ophthalmol 2015; 133: 770-777
- 158 Mavrikakis I, Rootman J. Diverse clinical presentations of orbital sarcoid. Am J Ophthalmol 2007; 144: 769-775
- 159 Demirci H, Christianson MD. Orbital and adnexal involvement in sarcoidosis: analysis of clinical features and systemic disease in 30 cases. Am J Ophthalmol 2011; 151: 1074-1080 e1071
- 160 McNab AA. The 2017 Doyne Lecture: the orbit as a window to systemic disease. Eye (Lond) 2018; 32: 248-261
- 161 Pasadhika S, Rosenbaum JT. Ocular Sarcoidosis. Clin Chest Med 2015; 36: 669-683
- 162 Aluclu MU, Keklikci U, Guzel A. et al. Melkersson-Rosenthal syndrome with partial oculomotor nerve palsy. Ann Saudi Med 2008; 28: 135-137
- 163 Shapiro M, Peters S, Spinelli HM. Melkersson-Rosenthal syndrome in the periocular area: a review of the literature and case report. Ann Plast Surg 2003; 50: 644-648
- 164 Cocuroccia B, Gubinelli E, Annessi G. et al. Persistent unilateral orbital and eyelid oedema as a manifestation of Melkersson-Rosenthal syndrome. J Eur Acad Dermatol Venereol 2005; 19: 107-111
- 165 Pierre-Filho Pde T, Rocha EM, Natalino R. et al. Upper eyelid oedema in Melkersson-Rosenthal syndrome. Clin Exp Ophthalmol 2004; 32: 439-440
- 166 Khandpur S, Malhotra AK, Khanna N. Melkersson-Rosenthal syndrome with diffuse facial swelling and multiple cranial nerve palsies. J Dermatol 2006; 33: 411-414
- 167 Rawlings NG, Valenzuela AA, Allen LH. et al. Isolated eyelid edema in Melkersson-Rosenthal syndrome: a case series. Eye (Lond) 2012; 26: 163-166
- 168 Ramaswamy B, Singh R, Manusrut M. et al. Sclerosing lipogranuloma of the eyelid: unusual complication following nasal packing in endoscopic sinus surgery. BMJ Case Rep. 2015 2015.
- 169 Witschel H, Geiger K. Paraffin induced sclerosing lipogranuloma of eyelids and anterior orbit following endonasal sinus surgery. Br J Ophthalmol 1994; 78: 61-65
- 170 Herrmann B, Rozot P, Baylac F. et al. [Lipogranuloma of the orbit. Apropos of a case]. J Fr Ophtalmol 1996; 19: 780-784
- 171 AlGhafri L, Galindo-Ferreiro A, Maktabi A. et al. Idiopathic orbital lipogranuloma. Int J Ophthalmol 2017; 10: 494-496
- 172 Jakobiec FA, Rai R, Rashid A. et al. Bilateral Eyelid Pseudoptosis From Lipogranulomas of the Preaponeurotic Fat Pads. Ophthalmic Plast Reconstr Surg 2015; 31: e125-131
- 173 Paik JS, Cho WK, Park GS. et al. Eyelid-associated complications after autogenous fat injection for cosmetic forehead augmentation. BMC Ophthalmol 2013; 13: 32
- 174 Park JY, Kim N. Periorbital Lipogranuloma after Facial Autologous Fat Injection and Its Treatment Outcomes. Korean J Ophthalmol 2016; 30: 10-16
- 175 Cafaro G, Croia C, Argyropoulou OD. et al. One year in review 2019: Sjogren's syndrome. Clin Exp Rheumatol 2019; 37 (Suppl. 118) 3-15
- 176 Mathews PM, Hahn S, Hessen M. et al. Ocular complications of primary Sjogren syndrome in men. Am J Ophthalmol 2015; 160: 447-452 e441
- 177 Danlos FX, Daoued F, Seror R. et al. Orbital Myositis and Primary Sjogren Syndrome. J Rheumatol 2015; 42: 1536-1537
- 178 Tonami H, Matoba M, Kuginuki Y. et al. Clinical and imaging findings of lymphoma in patients with Sjogren syndrome. J Comput Assist Tomogr 2003; 27: 517-524
- 179 Luemsamran P, Rootman J, White VA. et al. The role of biopsy in lacrimal gland inflammation: A clinicopathologic study. Orbit 2017; 36: 411-418
- 180 Ohshima K, Matsuo N, Yokoe S. et al. [A case of lacrimal gland malignant lymphoma, associated with Sjogren's syndrome]. Nippon Ganka Gakkai Zasshi 1991; 95: 386-392
- 181 Carubbi F, Cipriani P, Marrelli A. et al. Efficacy and safety of rituximab treatment in early primary Sjogren's syndrome: a prospective, multi-center, follow-up study. Arthritis Res Ther 2013; 15: R172
- 182 Kimura T. On the unusual granulations combined with hyperplastic changes of lymphatic tissue. Trans Soc Pathol Jpn 1948; 37: 179-180
- 183 Buder K, Ruppert S, Trautmann A. et al. Angiolymphoid hyperplasia with eosinophilia and Kimura's disease - a clinical and histopathological comparison. J Dtsch Dermatol Ges 2014; 12: 224-228
- 184 Claros P, Fokouo V, Nyada F. et al. Kimura's disease of the orbit: A modern diagnostic challenge. Eur Ann Otorhinolaryngol Head Neck Dis 2017; 134: 287-289
- 185 Googe PB, Harris NL, Mihm MC. Kimura's disease and angiolymphoid hyperplasia with eosinophilia: two distinct histopathological entities. J Cutan Pathol 1987; 14: 263-271
- 186 Kodama T, Kawamoto K. Kimura's disease of the lacrimal gland. Acta Ophthalmol Scand 1998; 76: 374-377
- 187 Monzen Y, Kiya K, Nishisaka T. Kimura's Disease of the Orbit Successfully Treated with Radiotherapy Alone: A Case Report. Case Rep Ophthalmol 2014; 5: 87-91
- 188 Bullock JD, Bartley GB, Campbell RJ. et al. Necrobiotic xanthogranuloma with paraproteinemia. Case report and a pathogenetic theory. Ophthalmology 1986; 93: 1233-1236
- 189 Shams PN, Rasmussen SL, Dolman PJ. Adult-Onset Asthma Associated With Simultaneous Conjunctival, Eyelid, and Orbital Xanthogranulomatosis Responsive to Systemic Immunosuppression. Ophthalmic Plast Reconstr Surg 2015; 31: e162-163
- 190 Elner VM, Mintz R, Demirci H. et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthalmic Plast Reconstr Surg 2006; 22: 36-40
- 191 Efebera Y, Blanchard E, Allam C. et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk 2011; 11: 298-302
- 192 Cives M, Simone V, Rizzo FM. et al. Erdheim-Chester disease: a systematic review. Crit Rev Oncol Hematol 2015; 95: 1-11
- 193 Estrada-Veras JI, O'Brien KJ, Boyd LC. et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv 2017; 1: 357-366
- 194 Merritt H, Pfeiffer ML, Richani K. et al. Erdheim-Chester disease with orbital involvement: Case report and ophthalmic literature review. Orbit 2016; 35: 221-226
- 195 Manousaridis K, Casper J, Schittkowski MP. et al. [Erdheim-Chester disease of the orbit with compressive optic neuropathy]. Ophthalmologe 2010; 107: 266-269
- 196 Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D. et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996; 75: 157-169
- 197 Altman J, Winkelmann RK. Diffuse normolipemic plane xanthoma. Generalized xanthelasma. Arch Dermatol 1962; 85: 633-640
- 198 Zak IT, Altinok D, Neilsen SS. et al. Xanthoma disseminatum of the central nervous system and cranium. AJNR Am J Neuroradiol 2006; 27: 919-921
- 199 Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol 2011; 147: 459-464
- 200 Minkov M, Lehrnbecher T. S1-Leitlinie 025/015 „Langerhanszell-Histiozytose (LCH) im Kindesalter In. 2017
- 201 Bertelmann E, Minko N, Lohneis P. et al. Benigne Neoplasien der Orbita. Klin Monatsbl Augenheilkd 2011; 228: 1113-1130
- 202 Maccheron LJ, McNab AA, Elder J. et al. Ocular adnexal Langerhans cell histiocytosis clinical features and management. Orbit 2006; 25: 169-177
- 203 Esmaili N, Harris GJ. Langerhans Cell Histiocytosis of the Orbit: Spectrum of Disease and Risk of Central Nervous System Sequelae in Unifocal Cases. Ophthalmic Plast Reconstr Surg 2016; 32: 28-34
- 204 Vemuganti GK, Naik MN, Honavar SG. Rosai dorfman disease of the orbit. J Hematol Oncol 2008; 1: 7
- 205 Bothra N, Kaliki S, Gowrishankar S. et al. Isolated unilateral eyelid Rosai-Dorfman disease. Oman J Ophthalmol 2018; 11: 300-302
- 206 Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol 1969; 87: 63-70
- 207 Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 1990; 7: 19-73
- 208 McClellan SF, Ainbinder DJ. Orbital Rosai-Dorfman disease: a literature review. Orbit 2013; 32: 341-346
- 209 Ezeanosike E, Ezeanosike OB, Akpan SI. et al. Rhabdomyoblastic differentiation in rosai dorfman disease of the orbit in a 12-year-old male. Niger J Clin Pract 2018; 21: 1670-1673
- 210 Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood 2014; 123: 2924-2933
- 211 Goel R, Raut A, Agarwal A. et al. A Rare Presentation of Orbital Castleman's Disease. Case Rep Ophthalmol Med 2020; 2020: 1012759
- 212 Schrock A, Gutgemann I, Keiner S. [Castleman's disease in ear, nose, and throat practice]. HNO 2007; 55 (Suppl. 01) E29-32
- 213 Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman's disease: a systematic review of 404 published cases. Ann Surg 2012; 255: 677-684
- 214 van Rhee F, Wong RS, Munshi N. et al. Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 2014; 15: 966-974
- 215 Kesper C, Viestenz A, Siebolts U. et al. [Orbital Amyloidosis - Comparison of Two Different Clinical Courses]. Klin Monbl Augenheilkd 2020; 237: 35-40
- 216 Leibovitch I, Selva D, Goldberg RA. et al. Periocular and orbital amyloidosis: clinical characteristics, management, and outcome. Ophthalmology 2006; 113: 1657-1664
- 217 Mora-Horna ER, Rojas-Padilla R, Lopez VG. et al. Ocular adnexal and orbital amyloidosis: a case series and literature review. Int Ophthalmol 2016; 36: 281-298
- 218 Naxer S, Behnes CL, Schittkowski MP. [Amyloidosis--a rare differential diagnosis of an orbital tumour]. Klin Monbl Augenheilkd 2011; 228: 555-564
- 219 Ladner-Merz S, Muller-Ladner U. [Amyloidoses]. Z Rheumatol 2008; 67: 677-682 quiz 683
- 220 Khaira M, Mutamba A, Meligonis G. et al. The use of radiotherapy for the treatment of localized orbital amyloidosis. Orbit 2008; 27: 432-437
- 221 Eghbali A, Hassan S, Seehra G. et al. Ophthalmological findings in Gaucher disease. Mol Genet Metab 2019; 127: 23-27
- 222 Prakalapakorn SG, Proia AD, Yanovitch TL. et al. Ocular and histologic findings in a series of children with infantile pompe disease treated with enzyme replacement therapy. J Pediatr Ophthalmol Strabismus 2014; 51: 355-362
- 223 Slingerland NW, Polling JR, van Gelder CM. et al. Ptosis, extraocular motility disorder, and myopia as features of pompe disease. Orbit 2011; 30: 111-113
- 224 Winter AW, Salimi A, Ospina LH. et al. Ophthalmic manifestations of Gaucher disease: the most common lysosomal storage disorder. Br J Ophthalmol 2019; 103: 315-326
- 225 Yanovitch TL, Banugaria SG, Proia AD. et al. Clinical and histologic ocular findings in pompe disease. J Pediatr Ophthalmol Strabismus 2010; 47: 34-40
- 226 Wabbels B, Ali N, Kunz WS. et al. [Chronic progressive external ophthalmoplegia and Kearns-Sayre syndrome : interdisciplinary diagnosis and therapy]. Ophthalmologe 2008; 105: 550-556
- 227 Pfeiffer MJ. [Chronic Progressive External Ophthalmoplegia Ptosis: Problems with Diagnostics and Treatment]. Klin Monbl Augenheilkd 2018; 235: 31-33
- 228 Pitceathly RD, Morrow JM, Sinclair CD. et al. Extra-ocular muscle MRI in genetically-defined mitochondrial disease. Eur Radiol 2016; 26: 130-137
- 229 Rahman S, Blok RB, Dahl HH. et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996; 39: 343-351
- 230 McKelvie P, Infeld B, Marotta R. et al. Late-adult onset Leigh syndrome. J Clin Neurosci 2012; 19: 195-202
- 231 Lake NJ, Compton AG, Rahman S. et al. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol 2016; 79: 190-203
- 232 Han J, Lee YM, Kim SM. et al. Ophthalmological manifestations in patients with Leigh syndrome. Br J Ophthalmol 2015; 99: 528-535
- 233 Fichter N, Schittkowski M, Guthoff R. Erkrankungen der Tränendrüse. Ophthalmologe 2005; 102: 399-425
- 234 Moreiras J. Tumors of the lacrimal gland - Orbit – examination, diagnosis, mircrosurgery, pathology. 2004
- 235 Font R, Gamel J. Epithelial tumours of the lacrimal gland: an analysis of 265 cases.In Jakobiec F (ed) Ocular and adnexal tumours. Aesculapius, Birmingham. 1978
- 236 Riedel K, Markl A, Hasenfratz G. et al. Epithelial tumors of the lacrimal gland: Clinico-pathologic correlation and management. Neurosurg Rev 1990; 13: 289-298
- 237 Andreasen S, Esmaeli B, Holstein L. et al. An Update on Tumors of the Lacrimal Gland. Asia-Pac. J Ophthalmol 2017; 6: 159-172
- 238 Cates C, Manners R, Rose G. Pleomorphic adenoma of the lacrimal gland in a 10 year old girl. Br J Ophthalmol 2002; 86: 249-250
- 239 Faktorovich E, Crawford J, Char D. Benign mixed tumor (pleomorphic adenoma) of the lacrimal gland in a 6-year-old boy. Am J Ophthalmol 1996; 122: 446-447
- 240 Mercado G, Gunduz K, Shields C. et al. Pleomorphic adenoma of the lacrimal gland in a teenager. Arch Ophthalmol 1998; 116: 962-963
- 241 Ni C, Kuo P, Dryja T. Histopathological classification of 272 primary epithelial tumors of the lacrimal gland. Chin Med J 1992; 105: 481-485
- 242 Rose G, Wright J. Pleomorphic adenoma of the lacrimal gland. Br J Ophthalmol 1992; 76: 395-400
- 243 Stefko S, DiBernardo C, Green W. et al. Pleomorphic adenoma of the lacrimal gland with extensive calcification. Arch Ophthalmol 2004; 122: 778-780
- 244 Stupp T, Pavlidis M, Buchner T. Pleomorphic Adenoma of the Lacrimal Gland in a Child After Treatmentof Acute Lymphoblastic Leukemia. Arch Ophthalmol 2004; 122: 1538-1540
- 245 Bonavolontà G, Tranfa F, Staibano S. et al. Warthin Tumor of the Lacrimal Gland. Am J Ophthalmol 1997; 124: 857-858
- 246 Ghosh D, Ekka J. Adenolymphoma of the lacrimal gland: A very rare case report. Int J Ocul Oncol oculoplasty 2018; 4: 144-145
- 247 Bartoletti-Stella A, Salfi N, Ceccarelli C. et al. Mitochondrial DNA Mutations in Oncocytic Adnexal Lacrimal Glands of the Conjunctiva. ARCH OPHTHALMOL 2011; 129: 664-666
- 248 Calle C, Castillo I, Eagle R. et al. Oncocytoma of the Lacrimal Gland: Case Report and Review of the Literature. Orbit 2006; 25: 243-247
- 249 Cameron J, Barras C. Oncocytoma of the eyelid. ACTA OPHTHALMOL SCAND 2004; 83: 125
- 250 George E, Swanson P, Newman B. et al. Oculocutaneous oncocytic tumors: clinicopathologic and immunohistochemical study of 2 cases with literature review. Am J Dermatopathol 2007; 29: 279-285
- 251 Morand B, Bettega G, Bland V. et al. Oncocytoma of the eyelid: an aggressive benign tumor. Ophthalmology 1998; 105: 2220-2224
- 252 Shields C, Shields J. Tumors of the conjunctiva and cornea. Survey of Ophthalmology 2004; 49: 3-24
- 253 Shields C, Shields J, Arbizo V. Oncocytoma of the caruncle. Am J Ophthalmol 1986; 102: 315-319
- 254 Surakiatchanukul T, Sioufi K, Pointdujour-Lim R. et al. Caruncular Oncocytoma Mimicking Malignant Melanoma. Ocul Oncol Pathol 3: 320-323
- 255 Bolzoni A, Pianta L, Farina D. et al. Benign myoepithelioma of the lacrimal gland: report of a case. Eur Arch Otorhinolaryngol 2005; 262: 186-188
- 256 Font R, Garner A. Myoepithelioma of the lacrimal gland: report of a case with spindle cell morphology. Brit J Ophthalmol 1992; 76: 634-636
- 257 Grossniklaus H, Wojno T, Wilson M. et al. Myoepithelioma of the Lacrimal Gland. Arch Ophthalmol 1997; 115: 1588-1590
- 258 Heathcote J, Hurwitz J, Dardick I. A Spindle-Cell Myoepithelioma of the Lacrimal Gland. Arch Ophthalmol 1990; 108: 1135-1139
- 259 Nickol T, E R Kleinsasser O. Myoepitheliome der Parotis. Jahresversammlung der Deutschen Gesellschaft für HNO-Heilkunde. 1998
- 260 Bajaj M, Pushker N, Kashyap S. Cystadenoma of the lacrimal gland. Orbit 2002; 21: 301-305
- 261 Bernardini F, Devoto M. JO C. Epithelial tumors of the lacimal gland: an update. Curr Opin in Ophthalmol 2008; 19: 409-413
- 262 Silbert J, Braich P, Misra R. Basal cell cystadenoma of the lacrimal gland: diagnostic pitfalls of a basaloid pattern in lacrimal tumours. Internat Ophthalmol. 2011 DOI: 43-46
- 263 Wang L, Zhang SK, Ma Y. et al. Papillary cystadenoma of the parotid gland: A case report. World J Clin Cases 2019; 7: 366-372
- 264 Pfeiffer M, Yin V, Bell D. et al. Sclerosing Polycystic Adenosis of the Lacrimal Gland. Ophthalmology 2013; 120: 873
- 265 Speight P, Barrett A. Salivary gland tumours: diagnostic challenges and an update on the latest WHO classification. Diagn Histopathol 2020; 26: 147-158
- 266 Brunnmann R, Ro J, Ordonez N. et al. Extrapleural Solitary Fibrous Tumor: A Clinicopathologic Study of 24 Cases. Mod Pathol 1999; 12: 1034-1042
- 267 Mupas-Uy J, Kitaguchia Y, Takahashia Y. et al. Solitary Fibrous Tumor in the Lacrimal Gland Fossa: A Case Report. Case Rep Ophthalmol 2016; 7: 398-403
- 268 Okike N, Bernatz P, Woolner L. Localized mesothelioma of the pleura. Benign and malignant variants. J Thorac Cardiovasc Surg 1978; 75: 363-372
- 269 Scott I, Tanenbaum M, Rubin D. Solitary fibrous tumor of the lacrimal gland fossa. Ophthalmology 1996; 103: 1613-1618
- 270 Vallat-Decouvelaere A, Dry S, Fletcher C. Atypical and malignant solitary fibrous tumor in extrathoracic locations; evidence of their comparability to intra-thoracic tumors. Am J Surg Pathol 1998; 22: 1501-1511
- 271 Woo K, Suh Y, Kim Y. Solitary fibrous tumor of the lacrimal sac. Ophthalmic Plast Reconstr Surg 1999; 15: 450-453
- 272 Bielory BP, Turbin RE, Mirani NM. et al. Radiographic and histological findings in an atypical orbital myxoma. Ophthalmol Eye Dis 2011; 3: 55
- 273 Demirci H, Shields CL, Eagle RCJ. et al. Report of a conjunctival myxoma case and review of the literature. Arch Ophthalmol 2006; 124: 735-738
- 274 Kini Rao A, Nayal B. Conjunctival myxoma-a case report. Malays J Med Sci 2013; 20: 92-94
- 275 Parikh D, Mukherjee B. Lacrimal gland myxoma. Indian J Ophthalmol 2017; 65: 887-889
- 276 Rambhatla S, Subramanian N, Gangadhara Sundar JK. et al. Myxoma of the orbit. Indian J Ophthalmol 2003; 51: 85-87
- 277 Bajaj M, Pushker N, Fibrous SK. histiocytoma of the lacrimal gland. Ophthal Plast Reconstr Surg 2007; 23: 145-147
- 278 Andrew N, Coupland S, Pirbhai A. et al. Lymphoid hyperplasia of the orbit and ocular adnexa: A clinical pathologic review. Survey of Ophthalmology 2016; 61: 778-790
- 279 Shields J, Shields C, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions: the 2002 Montgomery lecture, part 1. Ophtalmology. 2004 DOI: 997-1008
- 280 Wittekind C. TNM-Klassifikation maligner Tumoren. Achte Auflage. Aufl. WILEY-VCH; 2017
- 281 Devoto M, Croxatto J. Primary cystadenocarcinoma of the lacrimal gland. Ophthalmology 2003; 110: 2006-2010
- 282 Takahira M, Nakamura Y, Shimizu S. et al. Carcinosarcoma of the Lacrimal Gland Arising From a Pleomorphic Adenoma. Am J Ophthalmol 2005; 140: 340-341
- 283 Daisuke Y, Toshiyuki O, Shuichi Y. Case of primary neuroendocrine carcinoma in lacrimal gland. Acta Ophthalmol 2013; 91: 252
- 284 Gess A, Silkiss R. A merkel cell carcinoma of the lacrimal gland. Ophthal Plast Reconstr Surg. 28. 11-13
- 285 El-Naggar A, Chan J, Grandis J. World Health organization classification of head and neck tumours. 2017 DOI:
- 286 Weis E, Rootman J, Joly T. et al. Epithelial lacrimal gland tumors: pathologic classification and current understanding. Arch Ophthalmol 2009; 127: 1016-1028
- 287 Gamel J, Font R. Adenoidcystic carcinoma of the lacrimal gland: the clinical significance of a basaoid histologic pattern. Human Pathol 1982; 13: 219-225
- 288 Friedrich R, Bleckmann V. Adenoid cystic carcinoma of the salivary and lacrimal glands origin: localization, classification, clinical pathological correlation, treatment results and long-term follow-up control in 84 patients. Anticancer Res 2003; 23: 937-940
- 289 Goldberg R. Intraarterial chemotherapy. A welcome new idea for the management of adenoidcystic carcinoma of the lacrimal gland. Arch Ophthalmol 1998; 116: 372-373
- 290 Hamper K, Lazar F, Dietel M. Prognostic factors for adenoid cystic carcinoma of the head and neck: a retrospectiv evaluation of 96 cases. J Oral Pathol Med 1990; 19: 101-107
- 291 Henderson J. In Orbital Tumors New York: Raven Press;. 1994: 323-342
- 292 Lee D, Campbell R, Waller R. A clinicopathologic study of primary adenoid cystic carcinoma of the lacrimal gland. Ophthalmology 1985; 92: 128-134
- 293 Strianese D, Baldi G, Staibano S. Expression of apoptosis-related markers in malignant epithelial tumors of the lacrimal gland and their relation to clinical outcome. Br J Ophthalmol 2007; 91: 1239-1243
- 294 Tse D, Benedetto P, Dubovy S. Clinical analysis oft he effect on intraarterial cytoreductive chemotherapy in the treatment of lacrimal gland andoid cystic carcinoma. Am J Ophthalmol 2006; 141: 44-53
- 295 Wright J, Rose G, Garner A. Primary malignant neoplasms of the lacrimal gland. Br J Ophthalmol 1992; 76: 401-407
- 296 Karcioglu Z. Orbital Tumors. Diagnosis and Management. New York: Springer; 2005
- 297 Ahmad S, Esmaeli B, Williams M. American Joint Committee on Cancer classification predicts outcome of patients with lacrimal gland adenoid cystic carcinoma. Ophthalmology 2009; 116: 1210-1215
- 298 GROUP IHANS. Cervical lymph node metastasis in adenoid cystic carcinoma of the sinonasal tract, nasopharynx, lacrimal glands and external auditory canal: a collective international review. J Laryngol Otol 2016; 130: 1093-1097
- 299 Xiao R, Sethi R, Feng A. et al. The role of elective neck dissection in patients with adenoid cystic carcinoma of the head and neck. Laryngoscope 2019; 129: 2094-2104
- 300 Bördlein I. Mit schweren Ionen gegen hartnäckige Tumoren. Deutsches Ärzteblatt 2004; 101: 1718-1720
- 301 Hayashi K, Koto M, Ikawa H. et al. Efficacy and safety of carbon-ion radiotherapy for lacrimal gland carcinomas with extraorbital extension: a retrospective cohort study. Oncotarget 2018; 9: 12932-12940
- 302 Lesueur P, Rapeaud E, De Marzi L. et al. Adenoid Cystic Carcinoma of the Lacrimal Gland: High Dose Adjuvant Proton Therapy to Improve Patients Outcomes. Front Oncol 2020; 10: 135
- 303 Wolkow N, Jakobiec F, Lee H. et al. Long-term Outcomes of Globe-Preserving Surgery With Proton Beam Radiation for Adenoid Cystic Carcinoma of the Lacrimal Gland. Am J Ophthalmol. 2018 195.
- 304 Shields J, Shields C, Freire J. Plaque radiotherapy for selected orbital malignancies: preliminary observations: hte 2002 Montgomery Lecture, part 2. Ophthal Plast Reconstr Surg 2003; 19: 91-95
- 305 Tyl J, Blank L, Koornneef L. Brachytherapy in orbital tumors. Ophthalmology 1997; 104: 1475-1479
- 306 Gündüz A, Yesiltas Y, Shileds C. Overview of benign and malignant lacrimal gland tumors. Curr Opin Ophthalmol 2018; 29: 458-468
- 307 Ashton N. Epithelial tumors of the lacrimal gland. Mod Probl Ophthalmol 1975; 14: 306-323
- 308 Perzin K, Jakobiec F, Lovolsi V. Lacrimal gland malignant mixed tumors (carcinomas arising in benign mixed tumors): a clinicopathologic study. Cancer 1980; 45: 2593-2606
- 309 Shields C, Shields J, Eagle R. Clinicopathologic review of 142 cases of lacrimal gland lesions. Ophthalmology 1989; 96: 431-435
- 310 Henderson J, Farrow G. Primary malignant mixed tumors of the lacrimal gland. Report of 10 cases. Ophthalmology 1980; 87: 466-475
- 311 Jakobiec F, Bilyk J, Font R. Ophthalmic Pathology. An Atlas and Textbook. 4th ed. Aufl. Philadelphia. WB Saunders; 1996
- 312 Chen A, Garcia J, Bucci M. et al. The role of postoperative radiation therapy in carcinoma ex pleomorphic adenoma of the parotid gland. Int J Radiat Oncol Biol Phys 2007; 67: 138-143
- 313 Takahira M, Minato H, Takahashi M. Cystic carcinoma ex pleomorphic adenoma of the lacrimal gland. Ophthal Plast Reconstr Surg 2007; 23: 407-409
- 314 Hwang S, Kim K. High-grade Mucoepidermoid Carci- noma of the Lacrimal Gland. Korean J Ophthalmol 2018; 32: 426-427
- 315 Heaps R, Miller N, Albert D. Primary adenocarcinoma of the lacrimal gland. Ophtalmology 1993; 100: 1856-1860
- 316 von Holstein S, Coupland S. , D B. Epithelial Tumors of the Lacrimal Gland: a clinical, histopathological, surgical, and oncological survey. Acta Ophthalmol 2013; 91: 195-206
- 317 Dennie T. Metastastic her-2 amplified lacrimal gland carcinoma with response to lapatinib treatment. Case Rep Oncol Med 2015; 2015: 1-3
- 318 Totuk O, Demir M, Yapicier O. et al. Low-Grade Mucoepidermoid Carcinoma of the Lacrimal Gland in a Teenaged Patient. Case Rep Ophthalmol Med. 2017
- 319 Eviatar J, Hornblass A. Mucoepidermoid carcinoma of the lacrimal gland: 25 cases and a review and update of the literature. Ophthal Plast Reconstr Surg 1993; 9: 170-181
- 320 Sofinski S, Brown B, Rao N. Mucoepidermoid carcinoma of the lacrimal gland. Case report and review of the literature. Ophthalmic Plast Reconstr Surg 1986; 2: 147-151
- 321 Thorvaldsson S, Beahrs O, Woolner L. et al. Mucoepidermoid tumors of the major salivary glands. Am J Surg 1970; 120: 432-438
- 322 Katz E, Rootman J, Dolman P. et al. Primary Ductal Adenocarcinoma of the Lacrimal Gland. Ophthalmology 1996; 103: 157-162
- 323 Yang H, Wu C, Tsai C. et al. Primary ductal adenocarcinoma of lacrimal gland: Two case reports and review of the literature. Taiwan J Ophthalmol 2018; 8: 42-48
- 324 Ishida M, Hotta M, Kushima R. et al. Case of ductal adenocarcinoma ex pleomorphic adenoma of the lacrimal gland. Rinsho Byori 2009; 57: 746-751
- 325 Andreasen S, Grauslund M, Heegaard S. Lacrimal gland ductal carcinomas: clinical, morphological and genetic characterization and implications for targeted treatment. Acta Ophthalmol 2017; 95: 299-306
- 326 Jaspers H, Verbist B, Schoffelen R. et al. Androgen Receptor–Positive Salivary Duct Carcinoma: A Disease Entity With Promising New Treatment Options. J Clin Oncol 2011; 29: 473-476
- 327 See T, Stålhammar G, Tang T. et al. Primary ductal adenocarcinoma of the lacrimal gland: A review and report of five cases. Survey of Ophthalmology 2020; 65: 371-380
- 328 Anesi A, Negrello S, Lucchetti D. et al. Clinical Management of Acinic Cell Carcinoma of the Lacrimal Gland. Case Rep Oncol 2019; 12: 777-790
- 329 De Rosa G, Zeppa P, Tranta F. Acinic Cell Carcinoma in a Lacrimal Gland: first case report. Cancer 1986; 57: 1988-1991
- 330 Jang J, Kie J, Lee S. Acinic cell carcinoma of the lacrimal gland with intracranial extension. Ophthalmic Plast Reconstr Surg 2001; 17: 454-457
- 331 Rosenbaum P, Mahadevia P, Goodman L. Acinic cell carcinoma of the lacrimal gland. Arch Ophthalmol 1995; 113: 781-785
- 332 Briscoe D, Mahmood S, Bonshek R. et al. Primary sebaceous carcinoma of the lacrimal gland. Br J Ophthalmol 2001; 85: 625-626
- 333 Harvey P, Parsons M, Rennie I. Primary sebaceous carcinoma of lacrimal gland: a previously unreported primary neoplasm. Eye 1994; 8: 592-595
- 334 Kiratli H, Tarlan B, Fırat P. Primary sebaceous carcinoma of the lacrimal gland. Orbit 2012; 31: 352-354
- 335 Park H, Choi S. Primary sebaceous carcinoma of lacrimal gland: A case report and review of literature. World J Clin Cases 2018; 6: 1194-1198
- 336 Yamamoto N, Mizoe J, Hasegawa A. et al. Primary sebaceous carcinoma of the lacrimal gland treated by carbon ion radiotherapy. Int J Clin Oncol 2003; 8: 386-390
- 337 Herrera G. Light microscopic, ultrastructural and immunocytochemical spectrum of malignant lacrimal and salsivary gland tumors, including malignant mixed tumors. Pathobiology 1990; 58: 312-322
- 338 Iida K, Shikishima K, Okido M. et al. A case of malignant myoepithelioma in the lacrimal gland. Nippon Ganka Gakkai Zasshi 2001; 105: 42-46
- 339 Larbcharoensub N, Pangpunyakulchai D, Aroonroch R. et al. Lacrimal myoepithelial carcinoma ex recurrent pleomorphic adenoma: A clinicopathological report and review of the literature. Mol Clin Oncol 2018; 8: 209-213
- 340 Nagao T, Sugano I, Ishida Y. Salivary gland mmalignant myoepithelioma: a clinicopathologic and immunohistochemical study of ten cases. Cancer 1998; 83: 1292-1299
- 341 Xu T, Liao Z, Tang J. et al. Myoepithelial carcinoma of the headand neck: A report of 23 casesand literature review. Cancer Treatment. Communications 2014; 2: 24-29
- 342 Mahdi Y, Azami M, Daoudi R. et al. Diagnostic pitfall: primary myoepithelial carcinoma of the lacrimal gland, case report and literature review. BMC Clinical Pathology 2018; 18: 1-7
- 343 Yu G, Ma D, Sun K. Myoepithelial carcinoma of the salivary glands: behavior and management. Chin Med J 2003; 116: 163-165
- 344 Soumarová R, Lovasová Z, Vaňková M. Radiotherapy in malignant myoepithelioma of the soft palate – case report. Case Rep Oncol 2009; 2: 116-120
- 345 Nieder C, Schneller F, Grosu A. Radiotherapy and chemotherapy for myoepithelioma of the sellar region. Strahlenther Onkol 2005; 181: 260-263
- 346 Fenton S, Srinivasan S, Harnett A. et al. Primary squamous cell carcinoma of the lacrimal gland. Eye 2003; 17: 424-425
- 347 Su G, Patipa M, Font R. Primary squamous cell carcinoma arising from an epithelium-lined cyst of the lacrimal gland. Ophthal Plast Reconstr Surg 2005; 21: 383-385
- 348 Hotta K, Arisawa T, Mito H. et al. Primary squamous cell carcinoma of the lacrimal gland. Clin Experiment Ophthalmol 2005; 33: 534-536
- 349 Bernardini F, Orcioni G, Croxatto J. Oncocytic carcinoma of the lacrimal gland in a patient with neurofibromatosis. Ophthal Plast Reconstr Surg 2010; 26: 486-488
- 350 Kalantzis G, Norris J, El-Hindy N. et al. Oncocytic adenocarcinoma of the lacrimal gland: an unusual presentation. Eye 2013; 27: 104-105
- 351 Selva D, Davis G, Dodd T. Polymorphous Low-Grade Adenocarcinoma of the Lacrimal Gland. ARCH OPHTHALMOL 2004; 122: 915-917
- 352 Bortz J, Zhang P, Eagle R. et al. Secretory Carcinoma of the Lacrimal Gland: A Rare Case Report. Ophthalmic Plast Reconstr Surg 2018; 34: 154-157
- 353 Hyrczaa M, Andreasen S, Melchiorc L. et al. Primary Secretory Carcinoma of the Lacrimal Gland: Report of a New Entity. Am J Ophthalmol 2018; 193: 178-183
- 354 Kadir S. Synovial Sarcoma involving lacrimal gland: A rare clinical entity. Int J Ocu Oncol Oculoplast. 1: 14–18
- 355 Likhvantseva V, Safonova T, Kuzmin K. Primary granulocytic sarcoma of lacrimal gland. Vestn Oftalmol 2016; 132: 82-89
- 356 Yilmaz A, Saydam G, Sahin F. et al. Granulocytic sarcoma: a systematic review. Am J Blood Res 2013; 3: 265-270
- 357 Rajabi M, Riazi H, Hosseini S. et al. Malignant Peripheral Nerve Sheath Tumor of Lacrimal Nerve: A Case Report. Orbit 2015; 34: 41-44
- 358 Stark A, Hugo H, Buhl R. et al. Tumoren peripherer Nerven. Dtsch Arztebl 2002; 99: 928-933
- 359 Gündüz K, Shields J, Eagle R.. Malignant Rhabdoid Tumor of the Orbit. Arch Ophthalmol 1998; 116: 243-246
- 360 Johnson L, Sexton F, Goldberg S. Poorly differentiated primary orbital sarcoma (presumed malignant rhabdoid tumor): radiologic and histopathologic correlation. Arch Ophthalmol 1991; 109: 1275-1278
- 361 Orphanet. Rhabdoidtumor. In: Orphanet; 2020
- 362 Rootman J, Damji K, Dimmick J. Malignant rhabdoid tumor of the orbit. Ophthalmology 1989; 96: 1650-1654
- 363 Walford N, Deferrai R, Slater R. Intraorbital rhabdoid tumour following bilateral retinoblastoma. Histopathology 1992; 20: 170-173
- 364 Niffenegger J, Jakobiec F, Shore J. et al. Adult extrarenal rhabdoid tumor of the lacrimal gland. Ophthalmology 1992; 99: 567-574
- 365 Ferry J, Fung C, Zukerberg L. et al. Lymphoma of the ocular adnexa: A study of 353 cases 2007; 31: 170-184
- 366 Rasmussen P, Ralfkiaer E, Prause J. Malignant lymphoma of the lacrimal gland. A nation-based study. Arch Ophthalmol 2011; 129: 1275-1280
- 367 Johnson T, Tse D, Byrne G. Ocular adnexal lymphoid tumors: a clinicopathologic and molecular genetic study of 77 patients. Ophthal Plast Reconstr Surg 1999; 15: 171-179
- 368 Polito E, Galieni P, Leccisotti A. Clinical and radiological presentation of 95 orbital lymphoid tumors. Graefes Arch Clin Exp Ophthalmol 1996; 234: 504-509
- 369 Aranow M. Ocular adnexal lymphoma: evidence-based treatment approach. Int Ophthalmol Clin 2015; 55: 97-109
- 370 König L, Stade R, Rieber J. Radiotherapy of indolent orbital lymphomas: two radiation concepts. Strahlenther Onkol 2016; 192: 414-421
- 371 de Leval L, Jaffe E. Lymphoma Classification. Cancer J 2020; 26: 176-185
- 372 Shields J, Shields C, Eagle R. Metastatic renal cell carcinoma to the palpebral lobe of the lacrimal gland. Ophthal Plast Reconstr Surg 2001; 17: 191-194
- 373 Fowler J, Terplan K. Fibroma of the Orbit. Arch Ophthalmol 1942; 28: 263-271
- 374 Mortada A. Fibroma of the orbit. Brit J Ophthal 1971; 55: 350
- 375 Rong A, Ulloa-Padilla J, Blessing N. et al. Subperiosteal fibroma of the orbit. Orbit 2018; 37: 378-380
- 376 Rabelo N, da Silva V, do Espírito Santo M. et al. Orbit ossifying fibroma – Case report and literature review. Surg Neurol Int 2020; 11: 35
- 377 Sheikh BY. Cranial aneurysmal bone cyst “with special emphasis on endovascular management”. Acta Neurochir (Wien) 1999; 141: 601-610 discussion 610-601
- 378 Glien A, Meyer B, Wernert N. et al. [Orbital aneurysmal bone cyst in a 2-year-old child]. HNO 2007; 55: 281-286
- 379 Wang JC, Zhang M, Zhao XX. Aneurysmal bone cyst of the orbit. Chin Med J (Engl) 2015; 128: 562-563
- 380 Yu JW, Kim KU, Kim SJ. et al. Aneurysmal bone cyst of the orbit : a case report with literature review. J Korean Neurosurg Soc 2012; 51: 113-116
- 381 Ruiter DJ, van Rijssel TG, van der Velde EA. Aneurysmal bone cysts: a clinicopathological study of 105 cases. Cancer 1977; 39: 2231-2239
- 382 Biesecker JL, Marcove RC, Huvos AG. et al. Aneurysmal bone cysts. A clinicopathologic study of 66 cases. Cancer 1970; 26: 615-625
- 383 Schajowicz F. Tumors and tumorlike lesions of bone 2. Aufl. Berlin-Heidelberg-New York: Springer-Verlag; 1994
- 384 Oliveira AM, Chou MM. USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum Pathol 2014; 45: 1-11
- 385 Fay A, Dolman PJ. Laboratory Serologic Investigations.In Fay A, Dolman PJ , Hrsg. Diseases and Disorders of the Orbit and Ocular Adnexa E-book. Elsevier; 2017: 314
- 386 Mallen JR, Caranfa JT, Zimmerman D. et al. Atypical presentation of a solid-variant orbital aneurysmal bone cyst with a literature review. Neurochirurgie 2018; 64: 431-433
- 387 Chawla B, Khurana S, Kashyap S. Giant cell reparative granuloma of the orbit. Ophthalmic Plast Reconstr Surg 2013; 29: e94-95
- 388 Kamoshima Y, Sawamura Y, Imai T. et al. Giant cell tumor of the frontal bone in a girl: case report. Neurol Med Chir (Tokyo) 2011; 51: 798-800
- 389 Smolarz-Wojnowska A, Essig H, Gellrich NC. et al. [Orbital tumours in children and adolescents. Differential diagnostics and clinical symptoms]. Ophthalmologe 2010; 107: 543-548
- 390 Vernet O, Ducrey N, Deruaz JP. et al. Giant cell tumor of the orbit. Neurosurgery 1993; 32: 848-851
- 391 Yip CM, Lee HP, Hsu SS. et al. Left orbital roof giant cell tumor of bone: A case report. Surg Neurol Int 2018; 9: 127
- 392 Yamaguchi T, Dorfman HD. Giant cell reparative granuloma: a comparative clinicopathologic study of lesions in gnathic and extragnathic sites. Int J Surg Pathol 2001; 9: 189-200
- 393 de Lange J, van den Akker HP, van den Berg H. Central giant cell granuloma of the jaw: a review of the literature with emphasis on therapy options. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007; 104: 603-615
- 394 Auclair PL, Cuenin P, Kratochvil FJ. et al. A clinical and histomorphologic comparison of the central giant cell granuloma and the giant cell tumor. Oral Surg Oral Med Oral Pathol 1988; 66: 197-208
- 395 de Lange J, van Rijn RR, van den Berg H. et al. Regression of central giant cell granuloma by a combination of imatinib and interferon: a case report. Br J Oral Maxillofac Surg 2009; 47: 59-61
- 396 Roman-Romero L, Gonzalez-Garcia R. Cholesterol granuloma of the orbit. Report of cases and analysis of controversial treatment. J Maxillofac Oral Surg 2011; 10: 166-169
- 397 Loeffler KU, Kommerell G. Cholesterol granuloma of the orbit--pathogenesis and surgical management. Int Ophthalmol 1997; 21: 93-98
- 398 Selva D, Phipps SE, O'Connell JX. et al. Pathogenesis of orbital cholesterol granuloma. Clin Exp Ophthalmol 2003; 31: 78-82
- 399 Daus W, Voges J, Schwechheimer K. et al. [Cholesterol granuloma of the orbits--clinicopathologic study]. Klin Monbl Augenheilkd 1988; 193: 195-199
- 400 Hsu HT, Liao WC, Wu CC. et al. Cholesterol granuloma of the orbit. Kaohsiung J Med Sci 2017; 33: 422-423
- 401 Rong AJ, Erickson BP, Blessing NW. et al. Orbital cholesterol granuloma: A report and discussion of orbital findings. Am J Ophthalmol Case Rep 2019; 15: 100468
- 402 Arat YO, Chaudhry IA, Boniuk M. Orbitofrontal cholesterol granuloma: distinct diagnostic features and management. Ophthalmic Plast Reconstr Surg 2003; 19: 382-387
- 403 Aferzon M, Millman B, O'Donnell TR. et al. Cholesterol granuloma of the frontal bone. Otolaryngol Head Neck Surg 2002; 127: 578-581
- 404 Kabra R, Patel S, Shanbhag S. Orbital Chondroma: A rare mesenchymal tumor of orbit. Indian J Ophthalmol 2015; 63: 551-554
- 405 Garrity J, Henderson J. Henderson’s Orbital Tumors.In 4th ed. Aufl. USA: Lippincott Williams and Wilkis; 2007: 103–104
- 406 Rootman J, Connell J. Primary bone tumors of orbit. Surv Ophthalmol 2004; 49: 328-342
- 407 Shields J, Bakewell B, Augsburger J. et al. Classification and incidence of space-occupying lesions of the orbit. A survey of 645 biopsies. Arch Ophthalmol 1984; 102: 1606-1611
- 408 Pushker N, Meel R, Sharma S. et al. Giant orbital Schwannoma with fluid-fluid-levels. Br J Ophthalmol 2010; 95: 1168
- 409 Misra S, Gogri P, Misra N. et al. Recurrent neurofibroma of the orbit. Australas Med J 2013; 30: 189-191
- 410 Lee L, Gigantelli J, Kincaid M. Localized neurofibroma of the orbit: a radiographic and histopathologic study. Ophthal Plast Reconstr Surg 2000; 16: 241-246
- 411 Turbin R, Thompson C, Kennerdell J. et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophtalmology 2002; 109: 890-899
- 412 Schittkowski M, Fichter N., Orbitale Neoplasien Teil I. Einführung, vaskuläre und neurogene Neoplasien. Ophthalmologe 2012; 109: 1033-1046
- 413 Jain D, Ebrahimi K, Miller N. et al. Intraorbital meningiomas: a pathologic review using current World Health Organization criteria. Arch Pathol Lab Med 2010; 134: 766-770
- 414 Eddleman C, Liu J. Optic nerve sheath meningioma: current diagnosis and treatment. Neurosurg Focus 2007; 23: E4
- 415 Saeed P, Rootman J, Nugent R. et al. Optic nerve sheath meningiomas. Ophthalmology 2003; 110: 2019-2030
- 416 Ramey W, Arnold S, Chiu A. et al. A Rare Case of Optic Nerve Schwannoma: Case Report and Review of the Literature. Cureus 2015; 7: e265
- 417 Gnekow A. Low-grade chiasmatic-hypothalamic glioma-carboplatin and vincristine chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy - report from the multicenter treatment study for children and adolescents with a low grade glimoa - HIT-LGG 1996 - of the Society of Paediatric Oncology and Hematology (GPOH). Klin Padiatr 2004; 216: 331-342
- 418 Sharif S. Second primary tumors in neurofibromatosis 1 patients treated for optic nerve glioma: substantial risks after radiotherapy. J Clin Oncol 2006; 24: 2570-2575
- 419 Wilhelm H. Primary optic nerve tumors. Curr Opin Neurol 2009; 22: 11-18
- 420 Emesz M, Arlt E, Krall E. et al. Granularzelltumor der Orbita. Der. Ophthalmologe 2014; 111: 866-870
- 421 Huang N, Rayess H, Svider P. et al. Orbital Paraganglioma: A Systematic Review. J Neurol Surg B Skull Base 2017; 79: 407-412
- 422 Makhdoomi R, Nayil K, Santosh V. et al. Orbital paraganglioma--a case report and review of the literature. Clin Neuropathol 2010; 29: 100-104
- 423 Al-Khiary H, Ayoubi A, Elkhamary S. Primary orbital ganglioneuroma in a 2-year-old healthy boy. Saudi J Ophthalmol 2010; 24: 101-104
- 424 Cannon T, Brown H, Hughes B. Orbital Ganglioneuroma in a Patient With Chronic Progressive Proptosis. Arch Ophthalmol 2004; 122: 1712-1714
- 425 Hatsukawa Y, Furukawa A, Kawamura H. et al. Rhabdomyoma of the orbit in a child. Am J Ophthalmol 1997; 123: 142-144
- 426 Li Y, Li N, Zang W. et al. Rhabdomyoma of the orbit. J Pediatr Ophthalmol Strabismus 2008; 45: 113-115
- 427 Myung J, Kim I, Chun J et al. Rhabdomyoma of the orbit: a case report. Pediatric Radiolog. y. 32: 589–592
- 428 Gündüz K, Günalp I, Erden E. et al. Orbital leiomyoma: report of a case and review of the literature. Surv Ophthalmol 2004; 49: 237-242
- 429 Merani R, Khannah G, Mann S. et al. Orbital leiomyoma: a case report with clinical, radiological and pathological correlation. Clin Exp Ophthalmol 2005; 33: 408-411
- 430 Shah N, Chang W, White V. et al. Orbital lipoma: 2 cases and review of literature. Ophthalmic Plast Reconstr Surg 2007; 23: 202-205
- 431 Franchi G, Sleilati E, Soupre V. et al. Melanotic neuroectodermal tumour of infancy involving the orbit and maxilla: surgical management and follow-up strategy. Br J Plast Surg 2002; 55: 526-529
- 432 Koob M, Fayard C, Pariente D. et al. Prenatal diagnosis of orbital melanotic neuroectodermal tumor in infancy. Ultrasound Obstet Gynecol 2015; 46: 247-252
- 433 Soles B, Wilson A, Lucas D. et al. Melanotic Neuroectodermal Tumor of Infancy. Arch Pathol Lab Med 2018; 142: 1358-1363
- 434 NIH. Childhood Rhabdomyosarcoma Treatment (PDQ®)–Health Professional Version. In: National Cancer Institute at the National Institutes of Health; 2020
- 435 Al-Bargi H, Al-Maiman M. Orbital fibrosarcoma: a case report and literature review. Egypt J Oral Maxillofac Surg 2012; 3: 45-48
- 436 Bahrami A, Folpe A. Adult-type fibrosarcoma: A reevaluation of 163 putative cases diagnosed at a single institution over a 48-year period. Am J Surg Pathol 2010; 34: 1504-1513
- 437 Scruggs B, Ho S, Valenzuela A. Diagnostic challenges in primary orbital fibrosarcoma: a case report. Clin Ophthalmol 2014; 8: 2319-2323
- 438 Almater A, Alfaleh A, Alshomar K et al. Retinoblastoma: Update on Current Management, Retinoblastoma - Past, Present and Future. In Alkatan H, Hrsg.Retinoblastoma: IntechOpen; 2019
- 439 Cassoux N, Lumbroso L, Levy-Gabriel C. et al. Retinoblastoma: Update on Current Management. Asia-Pac. J Ophthalmol 2017; 6: 290-295
- 440 WHO. RETINOBLASTOMA - 2014 Review of Cancer Medicines on the WHO List of Essential Medicines. In: Union for International Cancer Control; 2014
- 441 Caignard A, Faguer R, Mercier P. et al. Optic nerve and visual pathways primary glioblastoma treated with radiotherapy and temozolomide chemotherapy. Eur J Ophthalmol 2014; 24: 637-640
- 442 Waheed S, Huang B, Zamora C. Glioblastoma involving the optic nerve. Am J Neuroradiol 2018; 41: e
- 443 Alireza M, Amelot A, Chauvet D. Poor prognosis and challenging treatment of optic nerve malignant gliomas: Literature review and case report series. World Neurosurg 2017; 97: e1-e6
- 444 Ashour O, Stalling M, Ramsey J. et al. Intraocular Medulloepithelioma. RadioGraphics 2018; 38: 194-199
- 445 Verdijk R. On the Classification and Grading of Medulloepithelioma of the Eye. Ocul Oncol Pathol 2016; 2: 190-193
- 446 Hei Y, Kang L, Yang X. Orbital alveolar soft part sarcoma: A report of 8 cases and review of the literature. Oncol Lett 2018; 15: 304-314
- 447 Inci E, Korkut N, Erem M. et al. [Alveolar soft tissue sarcoma]. HNO 2004; 52: 145-149
- 448 Rose A, Kabiru J, Rose G. Alveolar soft-part sarcoma of the orbit. Afr J Paediatr Surg 2011; 8: 82-84
- 449 Dirksen U, Jürgens H. S1-Leitlinie: Ewing-Sarkome des Kindes- und Jugendalters. Stand 06/2014, DOI: AWMF-Register Nr. 025/006
- 450 Kaliki S, Rathi S, Palkonda V. Primary orbital Ewing sarcoma family of tumors: a study of 12 cases. Eye 2018; 32: 615-621
- 451 Chaugule S, Putambekar A, Gavade S. et al. Primary Orbital Leiomyosarcoma in an Adult Male. Ophthal Plast Reconstr Surg 2019; 35: e27-e29
- 452 Klippenstein K, Wesley R, Glick A. Orbital Leiomyosarcoma After Retinoblastoma. Ophthalmic Surg Lasers Imaging. Retina 1999; 30: 579-583
- 453 Yeniad B, Tuncer S, Peksayar G. et al. Primary Orbital Leiomyosarcoma. Ophth. Plast Reconstr Surg 2009; 25: 154-155
- 454 Cai Y, McMenamin E, Geoff R. et al. Primary liposarcoma of the orbit: A clinicopathologic study of seven cases. Ann Diagn Pathol 2001; 5: 255-266
- 455 Garrity J, Henderson J, Cameron J. Henderson's Tumors of Primitive Mesoderm, Striated muscle, Adipose Tissue and Smooth muscle.In Garrity JA HJ, Cameron JD , Hrsg. Henderson's orbital tumors. 4th edition. AuflPhiladelphia: Lippincott Williams and Wilkins; 2007: 123–141
- 456 Alkatan H, Chaudhry I, Al-Qahtani A. Epithelioid sarcoma of the orbit. Ann Saudi Med 2011; 31: 187-189
- 457 Inazawa N, Hatakeyama N, Hori T. et al. Primary orbital neuroblastoma in a 1-month-old boy. Pediatr Int 2014; 56: 122-125
- 458 Vallinayagam M, Rao V, Pandian D. et al. Primary orbital neuroblastoma with intraocular extension. Indian J Ophthalmol 2015; 63: 684-686
- 459 Zhang N, Lin L. Presumed primary orbital neuroblastoma in a 20-month-old female. Ophthal Plast Reconstr Surg 2010; 26: 383-385
- 460 Rose A, Luthert P, Jayasena C. et al. Primary Orbital Melanoma: Presentation, Treatment, and Long-term Outcomes for 13 Patients. Front Oncol 2017; 7: 316
- 461 Kiratli H, Balci K, Güler G. Primary orbital endodermal sinus tumor (yolk sac tumor). J AAPOS 2008; 12: 623-625
- 462 Mogaddam A, Memar B, Aledavood A. et al. Isolated Orbital Endodermal Sinus Tumor. Ophthal Plast Reconstr Surg 2007; 23: 477-479
- 463 Eckardt A, Lemound J, Rana M. et al. Orbital lymphoma: diagnostic approach and treatment outcome. World J Surg Oncol. 2013 DOI: 73
- 464 Olsen T, Heegaard S. Orbitla Lymphoma. Surv Ophthalmol 2019; 64: 45-66
- 465 Sharma T, Kamath M. Diagnosis and Management of Orbital Lymphoma. Ophthalmic Pearls. 2015 DOI: 37-39
- 466 Bidar M, Wilson M, Laquis S. et al. Clinical and imaging characteristics of orbital leukemic tumors. Ophthalmic Plast Reconstr Surg 2007; 23: 87-93
- 467 Hatton M, Rubin P. Chronic Lymphocytic Leukemia of the Orbit. Arch Ophthalmol 2002; 120: 990-991
- 468 Maka E, Lukáts O, Tóth J. et al. Orbital tumour as initial manifestation of acute myeloid leukemia: granulocytic sarcoma: case report. Pathol Oncol Res 2008; 14: 209-211
- 469 Taylor C, Taylor R, Kinsey S. LEUKEMIC INFILTRATION OF THE ORBIT: Report of Three Cases and Literature Review. Pediatr Hematol Oncol 2005; 22: 415-422
- 470 Pausch N, Sterker I, Bauer U. Maligner intraorbitaler Tumor: Primum oder Metastase?. HNO 2016; 64: 262-264
- 471 Schenke T, Lehmann G, Kunze C. et al. Eine abszessverdächtige Raumforderung in der extraokulären Augenmuskulatur. HNO. 2020
- 472 Suarez C, Ferlito A, Lund V. Management of the orbit in malignant sinunasal tumors. Head Neck 2008; 30: 242-250
- 473 Evert K, Kühnel T, Weiß K. et al. Diagnostik und Management von Gefäßanomalien. Der. Pathologe 2019; 40: 422-430
- 474 Müller-Wille R, Wildgruber M, Sadick M. et al. Vascular Anomalies (Part II): Interventional Therapy of Peripheral Vascular Malformations. Fortschr Röntgenstr 2018; 190: 927-937
- 475 Sadick M, Müller-Wille R, Wildgruber M. et al. Vascular Anomalies (Part I): Classification and Diagnostics of Vascular Anomalies. Fortschr Röntgenstr 2018; 190: 825-835
- 476 Wassef M, Blei F, Adams D. et al. Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics 2015; 136: e203-e214
- 477 Fay A, Nguyen J, Jakobiec F. et al. Propranolol for Isolated Orbital Infantile Hemangioma. ARCH OPHTHALMOL 2010; 128: 256-258
- 478 Grantzow R, Schmittenbecher P, Höger P. et al. S2k-Leitlinie 006/100: Infantile Hämangiome im Säuglings- und Kleinkindesalter. AWMF online. 2015 DOI: 1-13
- 479 Goldsmith J, van de Rijn M, Syed N. Orbital Hemangiopericytoma and Solitary Fibrous Tumor: A Morphologic Continuum. Internat J Surg Pathol 2001; 9: 295-302
- 480 Pacheco L, Fernandes B, Miyamoto C. et al. Rapid growth of an orbital hemangiopericytoma with atypical histopathological findings. Clin Ophthalmol 2014; 8: 31-33
- 481 Westerfeld C, Allen C, Mihm M. et al. Non-Involuting Congenital Hemangioma Presenting as an Orbital Mass in a Newborn. Investigative Ophthalmology & Visual Science 48: 3582
- 482 Gbolahan O, Fasina O, Adisa A. et al. Spindle cell hemangioma: Unusual presentation of an uncommon tumor. J Oral Maxillofac Pathol 2015; 19: 406
- 483 Budimir I, Demirović A, Iveković R. et al. Epithelioid hemangioma of the orbit: case report. Acta Clin Croat 2015; 54: 92-95
- 484 McEachren T, Brownstein S, Jordan D. et al. Epithelioid hemangioma of the orbit. Ophtalmology 2000; 107: 806-810
- 485 Stiefel H, Ng J, Wilson D. et al. Orbital Cellular Epithelioid Hemangioma. Ocul Oncol Pathol 2019; 5: 424-431
- 486 Kamano H, Noguchi T, Yoshiura T. et al. Intraorbital lobular capillary hemangioma (pyogenic granuloma). Radiat Med 2008; 26: 609-612
- 487 Häußler S, Uecker F, Knopke S. et al. Tufted angiomas of the head and neck. HNO 2018; 66: S1-S6
- 488 Benesch M, Lackner H, Sorantin E. et al. Medikamentöse Therapie vaskulärer Anomalien. Paediatr Paedolog 2020; 55: 21-27
- 489 Cho W, Kim S, Park S. et al. Intracranial kaposiform hemangioendothelioma: proposal of a new malignant variant. J Pediatr Ophthalmol Strabismus 2009; 3: 147-150
- 490 Li B, Li Y, Tian X. et al. Unusual multifocal intraosseous papillary intralymphatic angioendothelioma (Dabska tumor) of facial bones: a case report and review of literature. Diagn Pathol 2013; 8: 160
- 491 Collaco L, Goncalves M, Gomes L. Orbital Kaposi’s Sarcoma in Acquired Immunodeficiency Syndrome. Eur J Ophthalmol 2000; 10: 88-90
- 492 Siddens J, Fishman J, Jackson I. et al. Primary orbital angiosarcoma: a case report. Ophthalmic Plast Reconstr Surg 1999; 15: 454-459
- 493 Lyon D, Tang T, Kidder T. Epithelioid Hemangioendothelioma of the Orbital Bones. Ophtalmology 1992; 99: 1773-1778
- 494 ISSVA. ISSVA Classification of Vascular Anomalies ©2018. In: International Society for the Study of Vascular Anomalies
- 495 Wildgruber M, Sadick M, Muller-Wille R. et al. Vascular tumors in infants and adolescents. Insights Imaging 2019; 10: 30
- 496 Gilbert P, Dubois J, Giroux F. New Treatment Approaches to Arteriovenous Malformations. Semin Intervent Radiol 2017; 34: 258-271
- 497 Schobinger RA, Fontaine R. Periphere Angiodysplasien.
Korrespondenzadresse
Publikationsverlauf
Artikel online veröffentlicht:
30. April 2021
© 2021. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
Literatur
- 1 Poloschek CM, Lagreze WA, Ridder GJ. et al. [Clinical and neuroradiological diagnostics of orbital tumors]. Ophthalmologe 2011; 108: 510-518
- 2 Plontke SK, Glien A, Kisser U. et al. [Diseases and Surgery of the Orbit]. Laryngorhinootologie 2020; 99: 896-917
- 3 AWMF. Rekonstruktion von Orbitadefekten (S2e-Leitlinie), Registernummer: 007/099. In: Arbeitsgemeinschaft medizinisch wissenschaftlicher Fachgesellschaften (AWMF); 2013
- 4 Fay A, Dolman PJ. Laboratory Serologic Investigations.In Fay A, Dolman PJ , Hrsg. Diseases and Disorders of the Orbit and Ocular Adnexa E-book. Elsevier; 2017: 57–77
- 5 Bonavolonta G, Tranfa F, de Conciliis C. et al. Dermoid cysts: 16-year survey. Ophthalmic Plast Reconstr Surg 1995; 11: 187-192
- 6 Jordan DR, Spitellie P, Brownstein S. et al. Orbital cholesterol granuloma and cholesteatoma: significance of differentiating the two. Ophthalmic Plast Reconstr Surg 2007; 23: 415-417
- 7 Sherman RP, Rootman J, Lapointe JS. Orbital dermoids: clinical presentation and management. Br J Ophthalmol 1984; 68: 642-652
- 8 Shields JA, Kaden IH, Eagle RC. et al. Orbital dermoid cysts: clinicopathologic correlations, classification, and management. The 1997 Josephine E. Schueler Lecture. Ophthalmic Plast Reconstr Surg 1997; 13: 265-276
- 9 Delfini R, Missori P, Iannetti G. et al. Mucoceles of the paranasal sinuses with intracranial and intraorbital extension: report of 28 cases. Neurosurgery 1993; 32: 901-906 discussion 906
- 10 Heichel J, Struck HG, Hammer T. et al. [Pediatric acute dacryocystitis due to frontoethmoidal mucocele]. HNO 2019; 67: 458-462
- 11 Nicollas R, Facon F, Sudre-Levillain I. et al. Pediatric paranasal sinus mucoceles: etiologic factors, management and outcome. Int J Pediatr Otorhinolaryngol 2006; 70: 905-908
- 12 Holtmann C, Brachert M, Spaniol K. et al. [Dacryolith in a lacrimal duct]. Ophthalmologe 2016; 113: 244-247
- 13 Tanaboonyawat S, Idowu OO, Copperman TS. et al. Dacryops - A review. Orbit 2020; 39: 128-134
- 14 Junemann A, Holbach LM. [Epithelial giant inclusion cyst 50 years after enucleation without orbital implant]. Klin Monbl Augenheilkd 1998; 212: 127-128
- 15 Kalantzis GK, Verity DH, Rose GE. Periocular implantation cysts: a late complication of ophthalmic surgery. Eye (Lond) 2014; 28: 1004-1007
- 16 Gupta R, Seith A, Guglani B. et al. Congenital cystic eye: features on MRI. Br J Radiol 2007; 80: e137-140
- 17 Guthoff R, Klein R, Lieb WE. Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 2004; 242: 268-271
- 18 Kuchle HJ, Normann J, Lubbering I. [Congenital cystic eye]. Klin Monbl Augenheilkd 1986; 188: 239-241
- 19 Schittkowski MP, Guthoff RF. Injectable self inflating hydrogel pellet expanders for the treatment of orbital volume deficiency in congenital microphthalmos: preliminary results with a new therapeutic approach. Br J Ophthalmol 2006; 90: 1173-1177
- 20 Schittkowski MP, Guthoff RF. Systemic and ophthalmological anomalies in congenital anophthalmic or microphthalmic patients. Br J Ophthalmol 2010; 94: 487-493
- 21 Badtke G, Tost M. Anophthalmus congenitus.In Velhagen K , Hrsg. Der Augenarzt, Band XI. Leipzig: VEB Georg Thieme; 1986: 328–344
- 22 Badtke G, Tost M. Mikrophthalmus congenitus.In Velhagen K , Hrsg. Der Augenarzt, Band XI. Leipzig: VEB Georg Thieme; 1986: 312–328
- 23 Chaudhry IA, Arat YO, Shamsi FA. et al. Congenital microphthalmos with orbital cysts: distinct diagnostic features and management. Ophthalmic Plast Reconstr Surg 2004; 20: 452-457
- 24 Oguz H, Ozturk A, San I. Congenital nasolacrimal duct occlusion with clinical anophthalmos: a possible new association. Ophthalmic Genet 2003; 24: 181-185
- 25 Pejic M, Luecke K, Meoded A. et al. Pediatric Cephaloceles: A Multimodality Review. Appl Radiol 2020; 49: 26-32
- 26 Terry A, Patrinely JR, Anderson RL. et al. Orbital meningoencephalocele manifesting as a conjunctival mass. Am J Ophthalmol 1993; 115: 46-49
- 27 Vashisht S, Ghai S, Hatimota P. et al. Cystic lesions of the orbit : A CT spectrum. Indian Journal of Radiology and Imaging 2003; 13: 139
- 28 Modi P, Shah NA, Bhalodia JN. et al. ORBITAL TUMORS IN CHILDREN: A DESCRIPTIVE STUDY AT TERTIARY CARE CENTRE. The Journal of medical research 2013; 3: 362-366
- 29 Green WR, Zimmerman LE. Ectopic lacrimal gland tissue. Report of eight cases with orbital involvement. Arch Ophthalmol 1967; 78: 318-327
- 30 Scheiner AJ, Frayer WC, Rorke LB et al. Ectopic brain tissue in the orbit. Eye (Lond) 1999; 13: ( Pt 2) 251–254
- 31 Damato PJ, Damato FJ. Neonatal Orbital Teratoma. Br J Ophthalmol 1962; 46: 685-691
- 32 Herman TE, Vachharajani A, Siegel MJ. Massive congenital orbital teratoma. J Perinatol 2009; 29: 396-397
- 33 Sesenna E, Ferri A, Thai E. et al. Huge orbital teratoma with intracranial extension: a case report. J Pediatr Surg 2010; 45: e27-31
- 34 Duke-Elder S. Anomalies of the Bony Orbit. In: Duke-Elder S, Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964: 942-949
- 35 Exner S, Bogusch G, Sokiranski R. Cribra orbitalia visualized in computed tomography. Ann Anat 2004; 186: 169-172
- 36 Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol 2004; 88: 833-834
- 37 Duke-Elder S. Deformities of the Face.In Duke-Elder S , Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964; 1007–1035
- 38 Kawamoto HK. The kaleidoscopic world of rare craniofacial clefts: order out of chaos (Tessier classification). Clin Plast Surg 1976; 3: 529-572
- 39 Fearon JA. Rare craniofacial clefts: a surgical classification. J Craniofac Surg 2008; 19: 110-112
- 40 Tessier P. Anatomical classification facial, cranio-facial and latero-facial clefts. J Maxillofac Surg 1976; 4: 69-92
- 41 Heike CL, Hing AV, Aspinall CA. et al. Clinical care in craniofacial microsomia: a review of current management recommendations and opportunities to advance research. Am J Med Genet C Semin Med Genet 2013; 163C: 271-282
- 42 Dupont S, Catala M, Hasboun D. et al. Progressive facial hemiatrophy and epilepsy: a common underlying dysgenetic mechanism. Neurology 1997; 48: 1013-1018
- 43 Fea AM, Aragno V, Briamonte C. et al. Parry Romberg syndrome with a wide range of ocular manifestations: a case report. BMC Ophthalmol 2015; 15: 119
- 44 Duke-Elder S. Deformities of the Skull.In Duke-Elder S , Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964: 1035–1072
- 45 Freudlsperger C, Castrillón-Oberndorfer G, Hoffmann J. et al. Isolierte, nichtsyndromale Kraniosynostosen. Der MKG-Chirurg 2013; 6: 301-313
- 46 Governale LS. Craniosynostosis. Pediatr Neurol 2015; 53: 394-401
- 47 Kotrikova B, Muhling J. [Surgery of craniofacial deformities]. Klin Monbl Augenheilkd 2004; 221: 970-977
- 48 Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr 2017; 29: 622-628
- 49 Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet 1999; 36: 177-182
- 50 Dubourg C, Bendavid C, Pasquier L. et al. Holoprosencephaly. Orphanet J Rare Dis 2007; 2: 8
- 51 Lustig LR, Holliday MJ, McCarthy EF. et al. Fibrous dysplasia involving the skull base and temporal bone. Arch Otolaryngol Head Neck Surg 2001; 127: 1239-1247
- 52 Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis 2009; 4: 5
- 53 Lannon DA, Earley MJ. Cherubism and its charlatans. Br J Plast Surg 2001; 54: 708-711
- 54 Grass S, Welkoborsky HJ, Mobius H. et al. [Orbital complications]. HNO 2018; 66: 800-811
- 55 Lehnerdt G, Peraud A, Berghaus A. et al. [Orbital and intracranial complications of acute sinusitis. Diagnostics and therapy in children and adolescents]. HNO 2011; 59: 75-86 quiz 87-78
- 56 Stammberger H. [Complications of inflammatory paranasal sinus diseases including iatrogenically-induced complications]. Eur Arch Otorhinolaryngol Suppl 1993; 1: 61-102
- 57 Welkoborsky HJ, Grass S, Deichmuller C. et al. Orbital complications in children: differential diagnosis of a challenging disease. Eur Arch Otorhinolaryngol 2015; 272: 1157-1163
- 58 Hamed-Azzam S, AlHashash I, Briscoe D. et al. Rare Orbital Infections ~ State of the Art ~ Part II. J Ophthalmic Vis Res 2018; 13: 183-190
- 59 Rootman J. Diseases of the orbit.Second edition Aufl. Lippincott Williams & Wilkins; Philadelphia: 2003
- 60 Madge SN, Prabhakaran VC, Shome D. et al. Orbital tuberculosis: a review of the literature. Orbit 2008; 27: 267-277
- 61 Fay A, Dolman PJ. Diseases and Disorders of the Orbit and Ocular Adnexa Expert Consult. In Fay A, Dolman PJ (eds). Elsevier; 2017
- 62 Heichel J, Griebsch M, Siebolts U. et al. [Bilateral periocular necrotizing fasciitis with unilateral orbital involvement]. Ophthalmologe 2020; 117: 1121-1125
- 63 Huber L, Budjan J, Rotter N. et al. [Necrotizing fasciitis in the head and neck region-three case reports and a review of the literature]. HNO 2020; 68: 935-943
- 64 Amrith S, Hosdurga Pai V, Ling WW. Periorbital necrotizing fasciitis -- a review. Acta Ophthalmol 2013; 91: 596-603
- 65 Wong CH, Chang HC, Pasupathy S. et al. Necrotizing fasciitis: clinical presentation, microbiology, and determinants of mortality. J Bone Joint Surg Am 2003; 85: 1454-1460
- 66 Lazzeri D, Lazzeri S, Figus M. et al. Periorbital necrotising fasciitis. Br J Ophthalmol 2010; 94: 1577-1585
- 67 Wladis EJ, Levin F, Shinder R. Clinical Parameters and Outcomes in Periorbital Necrotizing Fasciitis. Ophthalmic Plast Reconstr Surg 2015; 31: 467-469
- 68 Federspil P. German Society of Oto-Rhino-Laryngology H, Neck S. [Guidelines: Antibiotic treatment of infections of the head and neck: consensus report on behalf of the presidency of the German Society of Oto-Rhino-Laryngology, Head and Neck Surgery edited by P. Federspil, Homburg/Saar]. . HNO 2009; 57: 377-394
- 69 Barrani M, Massei F, Scaglione M. et al. Unusual onset of a case of chronic recurrent multifocal osteomyelitis. Pediatr Rheumatol Online J 2015; 13: 60
- 70 Dagan O, Barak Y, Metzker A. Pyoderma gangrenosum and sterile multifocal osteomyelitis preceding the appearance of Takayasu arteritis. Pediatr Dermatol 1995; 12: 39-42
- 71 Huber AM, Lam PY, Duffy CM. et al. Chronic recurrent multifocal osteomyelitis: clinical outcomes after more than five years of follow-up. J Pediatr 2002; 141: 198-203
- 72 Hu AC, Ng WKY. Orbital Osteomyelitis in the Pediatric Patient. J Craniofac Surg 2021; 32: 206-209
- 73 Cornely OA, Alastruey-Izquierdo A, Arenz D. et al. Global guideline for the diagnosis and management of mucormycosis: an initiative of the European Confederation of Medical Mycology in cooperation with the Mycoses Study Group Education and Research Consortium. Lancet Infect Dis 2019; 19: e405-e421
- 74 Bakhshaee M, Bojdi A, Allahyari A. et al. Acute invasive fungal rhinosinusitis: our experience with 18 cases. Eur Arch Otorhinolaryngol 2016; 273: 4281-4287
- 75 van Burik JA, Hare RS, Solomon HF. et al. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42: e61-65
- 76 Songu M, Unlu HH, Gunhan K. et al. Orbital exenteration: A dilemma in mucormycosis presented with orbital apex syndrome. Am J Rhinol 2008; 22: 98-103
- 77 Nicolai P, Lombardi D, Tomenzoli D. et al. Fungus ball of the paranasal sinuses: experience in 160 patients treated with endoscopic surgery. Laryngoscope 2009; 119: 2275-2279
- 78 Johnson TE, Casiano RR, Kronish JW. et al. Sino-orbital aspergillosis in acquired immunodeficiency syndrome. Arch Ophthalmol 1999; 117: 57-64
- 79 Levin LA, Avery R, Shore JW. et al. The spectrum of orbital aspergillosis: a clinicopathological review. Surv Ophthalmol 1996; 41: 142-154
- 80 Siddiqui AA, Shah AA, Bashir SH. Craniocerebral aspergillosis of sinonasal origin in immunocompetent patients: clinical spectrum and outcome in 25 cases. Neurosurgery 2004; 55: 602-611 discussion 611-603
- 81 Nielsen EW, Weisman RA, Savino PJ. et al. Aspergillosis of the sphenoid sinus presenting as orbital pseudotumor. Otolaryngol Head Neck Surg 1983; 91: 699-703
- 82 Sivak-Callcott JA, Livesley N, Nugent RA. et al. Localised invasive sino-orbital aspergillosis: characteristic features. Br J Ophthalmol 2004; 88: 681-687
- 83 Lamoth F, Calandra T. Early diagnosis of invasive mould infections and disease. J Antimicrob Chemother 2017; 72: i19-i28
- 84 McCarthy M, Rosengart A, Schuetz AN. et al. Mold infections of the central nervous system. N Engl J Med 2014; 371: 150-160
- 85 Green WR, Font RL, Zimmerman LE. Asperillosis of the orbit. Report of ten cases and review of the literature. Arch Ophthalmol 1969; 82: 302-313
- 86 Denning DW. Therapeutic outcome in invasive aspergillosis. Clin Infect Dis 1996; 23: 608-615
- 87 Robert Koch-Institut. Infektionsepidemiologisches Jahrbuch meldepflichtiger Krankheiten für 2019 Berlin: Robert Koch-Institut; 2020
- 88 Gomez Morales A, Croxatto JO, Crovetto L. et al. Hydatid cysts of the orbit. A review of 35 cases. Ophthalmology 1988; 95: 1027-1032
- 89 Xiao A, Xueyi C. Hydatid cysts of the orbit in Xinjiang: a review of 18 cases. Orbit 1999; 18: 151-155
- 90 Kars Z, Kansu T, Ozcan OE. et al. Orbital echinococcosis. Report of two cases studied by computerized tomography. J Clin Neuroophthalmol 1982; 2: 197-199
- 91 Chtira K, Benantar L, Aitlhaj H. et al. The surgery of intra-orbital hydatid cyst: a case report and literature review. Pan Afr Med J 2019; 33: 167
- 92 Senyuz OF, Yesildag E, Celayir S. Albendazole therapy in the treatment of hydatid liver disease. Surg Today 2001; 31: 487-491
- 93 Pushker N, Bajaj MS, Balasubramanya R. Disseminated cysticercosis involving orbit, brain and subcutaneous tissue. J Infect 2005; 51: e245-248
- 94 Walrath JD, Lalin SC, Leib ML. Cysticercosis isolated to the orbit. Ophthalmic Plast Reconstr Surg 2003; 19: 243-244
- 95 Sotelo J, Escobedo F, Rodriguez-Carbajal J. et al. Therapy of parenchymal brain cysticercosis with praziquantel. N Engl J Med 1984; 310: 1001-1007
- 96 Nentwich MM, Pleyer U, Schaller UC. et al. [International ophthalmology and travel medicine]. Ophthalmologe 2016; 113: 83-94
- 97 Mombaerts I, Rose GE, Garrity JA. Orbital inflammation: Biopsy first. Surv Ophthalmol 2016; 61: 664-669
- 98 Ben Simon GJ, Yoon MK, Atul J. et al. Clinical manifestations of orbital mass lesions at the Jules Stein Eye Institute, 1999-2003. Ophthalmic Surg Lasers Imaging 2006; 37: 25-32
- 99 Nölle B, Both M. Idiopathische Orbitaentzündung.In Pleyer U , Hrsg. Entzündliche Augenerkrankungen. Berlin, Heidelberg: Springer- Verlag; 2016: 48–57
- 100 Swamy BN, McCluskey P, Nemet A. et al. Idiopathic orbital inflammatory syndrome: clinical features and treatment outcomes. Br J Ophthalmol 2007; 91: 1667-1670
- 101 Fujii H, Fujisada H, Kondo T. et al. Orbital pseudotumor: histopathological classification and treatment. Ophthalmologica 1985; 190: 230-242
- 102 Mombaerts I, Goldschmeding R, Schlingemann RO. et al. What is orbital pseudotumor?. Surv Ophthalmol 1996; 41: 66-78
- 103 Rootman J, Nugent R. The classification and management of acute orbital pseudotumors. Ophthalmology 1982; 89: 1040-1048
- 104 Rootman J, McCarthy M, White V. et al. Idiopathic sclerosing inflammation of the orbit. A distinct clinicopathologic entity. Ophthalmology 1994; 101: 570-584
- 105 Iaconetta G, Stella L, Esposito M. et al. Tolosa-Hunt syndrome extending in the cerebello-pontine angle. Cephalalgia 2005; 25: 746-750
- 106 Speth MM, Metternich FU, Mina R. et al. Tolosa-Hunt-Syndrom, das sich als migrierende, auf Kortikosteroid reagierende CN3- und CN6-Lähmung bei verschatteten Keilbeinhöhlen darstellt. forum hno: omnimed Verlag 05/2020;
- 107 Halabi T, Sawaya R. Successful Treatment of Tolosa-Hunt Syndrome after a Single Infusion of Infliximab. J Clin Neurol 2018; 14: 126-127
- 108 Mormont E, Laloux P, Vauthier J. et al. Radiotherapy in a case of Tolosa-Hunt syndrome. Cephalalgia 2000; 20: 931-933
- 109 Perez CA, Evangelista M. Evaluation and Management of Tolosa-Hunt Syndrome in Children: A Clinical Update. Pediatr Neurol 2016; 62: 18-26
- 110 Zhang X, Zhang W, Liu R. et al. Factors that influence Tolosa-Hunt syndrome and the short-term response to steroid pulse treatment. J Neurol Sci 2014; 341: 13-16
- 111 Espinoza GM, Liu JL. Orbital Vasculitides-Differential Diagnosis. Curr Rheumatol Rep 2019; 21: 54
- 112 Tiegs-Heiden CA, Eckel LJ, Hunt CH. et al. Immunoglobulin G4-related disease of the orbit: imaging features in 27 patients. AJNR Am J Neuroradiol 2014; 35: 1393-1397
- 113 Erdei A, Steiber Z, Molnar C. et al. Exophthalmos in a young woman with no graves' disease - a case report of IgG4-related orbitopathy. BMC Ophthalmol 2018; 18: 5
- 114 Jurkov M, Olze H, Klauschen F. et al. [IgG4-Related Orbitopathy as an Important Differential Diagnosis of Advanced Silent Sinus Syndrome. German version]. HNO 2020; 68: 864-868
- 115 Mulholland GB, Jeffery CC, Satija P. et al. Immunoglobulin G4-related diseases in the head and neck: a systematic review. J Otolaryngol Head Neck Surg 2015; 44: 24
- 116 Detiger SE, Karim AF, Verdijk RM. et al. The treatment outcomes in IgG4-related orbital disease: a systematic review of the literature. Acta Ophthalmol 2019; 97: 451-459
- 117 Plaza JA, Garrity JA, Dogan A. et al. Orbital inflammation with IgG4-positive plasma cells: manifestation of IgG4 systemic disease. Arch Ophthalmol 2011; 129: 421-428
- 118 Perumal B, Black EH, Levin F. et al. Non-infectious orbital vasculitides. Eye (Lond) 2012; 26: 630-639
- 119 Sfiniadaki E, Tsiara I, Theodossiadis P. et al. Ocular Manifestations of Granulomatosis with Polyangiitis: A Review of the Literature. Ophthalmol Ther 2019; 8: 227-234
- 120 Mukhtyar C, Flossmann O, Hellmich B. et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis 2008; 67: 1004-1010
- 121 Holle JU, Voigt C, Both M. et al. Orbital masses in granulomatosis with polyangiitis are associated with a refractory course and a high burden of local damage. Rheumatology (Oxford) 2013; 52: 875-882
- 122 Perry SR, Rootman J, White VA. The clinical and pathologic constellation of Wegener granulomatosis of the orbit. Ophthalmology 1997; 104: 683-694
- 123 Chang SY, Keogh KA, Lewis JE. et al. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener's): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol 2013; 44: 2432-2437
- 124 Martinez Del Pero M, Chaudhry A, Jones RB. et al. B-cell depletion with rituximab for refractory head and neck Wegener's granulomatosis: a cohort study. Clin Otolaryngol 2009; 34: 328-335
- 125 Takanashi T, Uchida S, Arita M. et al. Orbital inflammatory pseudotumor and ischemic vasculitis in Churg-Strauss syndrome: report of two cases and review of the literature. Ophthalmology 2001; 108: 1129-1133
- 126 Cottin V, Cordier JF. Churg-Strauss syndrome. Allergy 1999; 54: 535-551
- 127 Masi AT, Hunder GG, Lie JT. et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990; 33: 1094-1100
- 128 Vaglio A, Moosig F, Zwerina J. Churg-Strauss syndrome: update on pathophysiology and treatment. Curr Opin Rheumatol 2012; 24: 24-30
- 129 Guillevin L, Lhote F, Gayraud M. et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996; 75: 17-28
- 130 Nagel MA, White T, Khmeleva N. et al. Analysis of Varicella-Zoster Virus in Temporal Arteries Biopsy Positive and Negative for Giant Cell Arteritis. JAMA Neurol 2015; 72: 1281-1287
- 131 Borruat FX, Bogousslavsky J, Uffer S. et al. Orbital infarction syndrome. Ophthalmology 1993; 100: 562-568
- 132 Stone JH, Tuckwell K, Dimonaco S. et al. Trial of Tocilizumab in Giant-Cell Arteritis. N Engl J Med 2017; 377: 317-328
- 133 Seyahi E. Takayasu arteritis: an update. Curr Opin Rheumatol 2017; 29: 51-56
- 134 Tugal-Tutkun I. Systemic vasculitis and the eye. Curr Opin Rheumatol 2017; 29: 24-32
- 135 Misra DP, Wakhlu A, Agarwal V. et al. Recent advances in the management of Takayasu arteritis. Int J Rheum Dis 2019; 22 (Suppl. 01) 60-68
- 136 Iliescu DA, Timaru CM, Batras M. et al. Cogan's Syndrome. Rom J Ophthalmol 2015; 59: 6-13
- 137 Tayer-Shifman OE, Ilan O, Tovi H. et al. Cogan's syndrome--clinical guidelines and novel therapeutic approaches. Clin Rev Allergy Immunol 2014; 47: 65-72
- 138 Grasland A, Pouchot J, Hachulla E. et al. Typical and atypical Cogan's syndrome: 32 cases and review of the literature. Rheumatology (Oxford) 2004; 43: 1007-1015
- 139 Gluth MB, Baratz KH, Matteson EL. et al. Cogan syndrome: a retrospective review of 60 patients throughout a half century. Mayo Clin Proc 2006; 81: 483-488
- 140 Padoan R, Cazzador D, Pendolino AL. et al. Cogan's syndrome: new therapeutic approaches in the biological era. Expert Opin Biol Ther 2019; 19: 781-788
- 141 Kokturk A. Clinical and Pathological Manifestations with Differential Diagnosis in Behcet's Disease. Patholog Res Int 2012; 2012: 690390
- 142 Hammami S, Yahia SB, Mahjoub S. et al. Orbital inflammation associated with Behcet's disease. Clin Exp Ophthalmol 2006; 34: 188-190
- 143 Urruticoechea-Arana A, Cobo-Ibanez T, Villaverde-Garcia V. et al. Efficacy and safety of biological therapy compared to synthetic immunomodulatory drugs or placebo in the treatment of Behcet's disease associated uveitis: a systematic review. Rheumatol Int 2019; 39: 47-58
- 144 Bettiol A, Silvestri E, Di Scala G. et al. The right place of interleukin-1 inhibitors in the treatment of Behcet's syndrome: a systematic review. Rheumatol Int 2019; 39: 971-990
- 145 Hedrich CM, Schnabel A, Hospach T. Kawasaki Disease. Front Pediatr 2018; 6: 198
- 146 Demirsoy S, Gucuyener K, Olgunturk R. et al. A case of Kawasaki syndrome associated with preseptal cellulitis in orbita. Turk J Pediatr 1988; 30: 55-59
- 147 Felz MW, Patni A, Brooks SE. et al. Periorbital vasculitis complicating Kawasaki syndrome in an infant. Pediatrics 1998; 101: E9
- 148 Sheard RM, Pandey KR, Barnes ND. et al. Kawasaki disease presenting as orbital cellulitis. J Pediatr Ophthalmol Strabismus 2000; 37: 123-125
- 149 Son MB, Gauvreau K, Burns JC. et al. Infliximab for intravenous immunoglobulin resistance in Kawasaki disease: a retrospective study. J Pediatr 2011; 158: 644-649 e641
- 150 Read RW. Clinical mini-review: systemic lupus erythematosus and the eye. Ocul Immunol Inflamm 2004; 12: 87-99
- 151 Arthurs BP, Khalil MK, Chagnon F. et al. Orbital infarction and melting in a patient with systemic lupus erythematosus. Ophthalmology 1999; 106: 2387-2390
- 152 Nabili S, McCarey DW, Browne B. et al. A case of orbital myositis associated with rheumatoid arthritis. Ann Rheum Dis 2002; 61: 938-939
- 153 Sanchez IJ, Finger DR, Bradshaw DJ. Orbital inflammatory syndrome and systemic lupus erythematosus. J Clin Rheumatol 1995; 1: 295-298
- 154 Kokotis P, Theodossiadis P, Bouros C. et al. Bilateral ocular myositis as a late complication of dermatomyositis. J Rheumatol 2005; 32: 379-381
- 155 Lokdarshi G, Pushker N, Bajaj MS. Sclerosing Lesions of the Orbit: A Review. Middle East Afr J Ophthalmol 2015; 22: 447-451
- 156 Petrarolha SM, Rodrigues BS, Filho FD. et al. Unilateral Eyelid Edema as Initial Sign of Orbital Sarcoidosis. Case Rep Ophthalmol Med 2016; 2016: 6912927
- 157 Rosenbaum JT, Choi D, Wilson DJ. et al. Parallel Gene Expression Changes in Sarcoidosis Involving the Lacrimal Gland, Orbital Tissue, or Blood. JAMA Ophthalmol 2015; 133: 770-777
- 158 Mavrikakis I, Rootman J. Diverse clinical presentations of orbital sarcoid. Am J Ophthalmol 2007; 144: 769-775
- 159 Demirci H, Christianson MD. Orbital and adnexal involvement in sarcoidosis: analysis of clinical features and systemic disease in 30 cases. Am J Ophthalmol 2011; 151: 1074-1080 e1071
- 160 McNab AA. The 2017 Doyne Lecture: the orbit as a window to systemic disease. Eye (Lond) 2018; 32: 248-261
- 161 Pasadhika S, Rosenbaum JT. Ocular Sarcoidosis. Clin Chest Med 2015; 36: 669-683
- 162 Aluclu MU, Keklikci U, Guzel A. et al. Melkersson-Rosenthal syndrome with partial oculomotor nerve palsy. Ann Saudi Med 2008; 28: 135-137
- 163 Shapiro M, Peters S, Spinelli HM. Melkersson-Rosenthal syndrome in the periocular area: a review of the literature and case report. Ann Plast Surg 2003; 50: 644-648
- 164 Cocuroccia B, Gubinelli E, Annessi G. et al. Persistent unilateral orbital and eyelid oedema as a manifestation of Melkersson-Rosenthal syndrome. J Eur Acad Dermatol Venereol 2005; 19: 107-111
- 165 Pierre-Filho Pde T, Rocha EM, Natalino R. et al. Upper eyelid oedema in Melkersson-Rosenthal syndrome. Clin Exp Ophthalmol 2004; 32: 439-440
- 166 Khandpur S, Malhotra AK, Khanna N. Melkersson-Rosenthal syndrome with diffuse facial swelling and multiple cranial nerve palsies. J Dermatol 2006; 33: 411-414
- 167 Rawlings NG, Valenzuela AA, Allen LH. et al. Isolated eyelid edema in Melkersson-Rosenthal syndrome: a case series. Eye (Lond) 2012; 26: 163-166
- 168 Ramaswamy B, Singh R, Manusrut M. et al. Sclerosing lipogranuloma of the eyelid: unusual complication following nasal packing in endoscopic sinus surgery. BMJ Case Rep. 2015 2015.
- 169 Witschel H, Geiger K. Paraffin induced sclerosing lipogranuloma of eyelids and anterior orbit following endonasal sinus surgery. Br J Ophthalmol 1994; 78: 61-65
- 170 Herrmann B, Rozot P, Baylac F. et al. [Lipogranuloma of the orbit. Apropos of a case]. J Fr Ophtalmol 1996; 19: 780-784
- 171 AlGhafri L, Galindo-Ferreiro A, Maktabi A. et al. Idiopathic orbital lipogranuloma. Int J Ophthalmol 2017; 10: 494-496
- 172 Jakobiec FA, Rai R, Rashid A. et al. Bilateral Eyelid Pseudoptosis From Lipogranulomas of the Preaponeurotic Fat Pads. Ophthalmic Plast Reconstr Surg 2015; 31: e125-131
- 173 Paik JS, Cho WK, Park GS. et al. Eyelid-associated complications after autogenous fat injection for cosmetic forehead augmentation. BMC Ophthalmol 2013; 13: 32
- 174 Park JY, Kim N. Periorbital Lipogranuloma after Facial Autologous Fat Injection and Its Treatment Outcomes. Korean J Ophthalmol 2016; 30: 10-16
- 175 Cafaro G, Croia C, Argyropoulou OD. et al. One year in review 2019: Sjogren's syndrome. Clin Exp Rheumatol 2019; 37 (Suppl. 118) 3-15
- 176 Mathews PM, Hahn S, Hessen M. et al. Ocular complications of primary Sjogren syndrome in men. Am J Ophthalmol 2015; 160: 447-452 e441
- 177 Danlos FX, Daoued F, Seror R. et al. Orbital Myositis and Primary Sjogren Syndrome. J Rheumatol 2015; 42: 1536-1537
- 178 Tonami H, Matoba M, Kuginuki Y. et al. Clinical and imaging findings of lymphoma in patients with Sjogren syndrome. J Comput Assist Tomogr 2003; 27: 517-524
- 179 Luemsamran P, Rootman J, White VA. et al. The role of biopsy in lacrimal gland inflammation: A clinicopathologic study. Orbit 2017; 36: 411-418
- 180 Ohshima K, Matsuo N, Yokoe S. et al. [A case of lacrimal gland malignant lymphoma, associated with Sjogren's syndrome]. Nippon Ganka Gakkai Zasshi 1991; 95: 386-392
- 181 Carubbi F, Cipriani P, Marrelli A. et al. Efficacy and safety of rituximab treatment in early primary Sjogren's syndrome: a prospective, multi-center, follow-up study. Arthritis Res Ther 2013; 15: R172
- 182 Kimura T. On the unusual granulations combined with hyperplastic changes of lymphatic tissue. Trans Soc Pathol Jpn 1948; 37: 179-180
- 183 Buder K, Ruppert S, Trautmann A. et al. Angiolymphoid hyperplasia with eosinophilia and Kimura's disease - a clinical and histopathological comparison. J Dtsch Dermatol Ges 2014; 12: 224-228
- 184 Claros P, Fokouo V, Nyada F. et al. Kimura's disease of the orbit: A modern diagnostic challenge. Eur Ann Otorhinolaryngol Head Neck Dis 2017; 134: 287-289
- 185 Googe PB, Harris NL, Mihm MC. Kimura's disease and angiolymphoid hyperplasia with eosinophilia: two distinct histopathological entities. J Cutan Pathol 1987; 14: 263-271
- 186 Kodama T, Kawamoto K. Kimura's disease of the lacrimal gland. Acta Ophthalmol Scand 1998; 76: 374-377
- 187 Monzen Y, Kiya K, Nishisaka T. Kimura's Disease of the Orbit Successfully Treated with Radiotherapy Alone: A Case Report. Case Rep Ophthalmol 2014; 5: 87-91
- 188 Bullock JD, Bartley GB, Campbell RJ. et al. Necrobiotic xanthogranuloma with paraproteinemia. Case report and a pathogenetic theory. Ophthalmology 1986; 93: 1233-1236
- 189 Shams PN, Rasmussen SL, Dolman PJ. Adult-Onset Asthma Associated With Simultaneous Conjunctival, Eyelid, and Orbital Xanthogranulomatosis Responsive to Systemic Immunosuppression. Ophthalmic Plast Reconstr Surg 2015; 31: e162-163
- 190 Elner VM, Mintz R, Demirci H. et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthalmic Plast Reconstr Surg 2006; 22: 36-40
- 191 Efebera Y, Blanchard E, Allam C. et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk 2011; 11: 298-302
- 192 Cives M, Simone V, Rizzo FM. et al. Erdheim-Chester disease: a systematic review. Crit Rev Oncol Hematol 2015; 95: 1-11
- 193 Estrada-Veras JI, O'Brien KJ, Boyd LC. et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv 2017; 1: 357-366
- 194 Merritt H, Pfeiffer ML, Richani K. et al. Erdheim-Chester disease with orbital involvement: Case report and ophthalmic literature review. Orbit 2016; 35: 221-226
- 195 Manousaridis K, Casper J, Schittkowski MP. et al. [Erdheim-Chester disease of the orbit with compressive optic neuropathy]. Ophthalmologe 2010; 107: 266-269
- 196 Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D. et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996; 75: 157-169
- 197 Altman J, Winkelmann RK. Diffuse normolipemic plane xanthoma. Generalized xanthelasma. Arch Dermatol 1962; 85: 633-640
- 198 Zak IT, Altinok D, Neilsen SS. et al. Xanthoma disseminatum of the central nervous system and cranium. AJNR Am J Neuroradiol 2006; 27: 919-921
- 199 Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol 2011; 147: 459-464
- 200 Minkov M, Lehrnbecher T. S1-Leitlinie 025/015 „Langerhanszell-Histiozytose (LCH) im Kindesalter In. 2017
- 201 Bertelmann E, Minko N, Lohneis P. et al. Benigne Neoplasien der Orbita. Klin Monatsbl Augenheilkd 2011; 228: 1113-1130
- 202 Maccheron LJ, McNab AA, Elder J. et al. Ocular adnexal Langerhans cell histiocytosis clinical features and management. Orbit 2006; 25: 169-177
- 203 Esmaili N, Harris GJ. Langerhans Cell Histiocytosis of the Orbit: Spectrum of Disease and Risk of Central Nervous System Sequelae in Unifocal Cases. Ophthalmic Plast Reconstr Surg 2016; 32: 28-34
- 204 Vemuganti GK, Naik MN, Honavar SG. Rosai dorfman disease of the orbit. J Hematol Oncol 2008; 1: 7
- 205 Bothra N, Kaliki S, Gowrishankar S. et al. Isolated unilateral eyelid Rosai-Dorfman disease. Oman J Ophthalmol 2018; 11: 300-302
- 206 Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol 1969; 87: 63-70
- 207 Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 1990; 7: 19-73
- 208 McClellan SF, Ainbinder DJ. Orbital Rosai-Dorfman disease: a literature review. Orbit 2013; 32: 341-346
- 209 Ezeanosike E, Ezeanosike OB, Akpan SI. et al. Rhabdomyoblastic differentiation in rosai dorfman disease of the orbit in a 12-year-old male. Niger J Clin Pract 2018; 21: 1670-1673
- 210 Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood 2014; 123: 2924-2933
- 211 Goel R, Raut A, Agarwal A. et al. A Rare Presentation of Orbital Castleman's Disease. Case Rep Ophthalmol Med 2020; 2020: 1012759
- 212 Schrock A, Gutgemann I, Keiner S. [Castleman's disease in ear, nose, and throat practice]. HNO 2007; 55 (Suppl. 01) E29-32
- 213 Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman's disease: a systematic review of 404 published cases. Ann Surg 2012; 255: 677-684
- 214 van Rhee F, Wong RS, Munshi N. et al. Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 2014; 15: 966-974
- 215 Kesper C, Viestenz A, Siebolts U. et al. [Orbital Amyloidosis - Comparison of Two Different Clinical Courses]. Klin Monbl Augenheilkd 2020; 237: 35-40
- 216 Leibovitch I, Selva D, Goldberg RA. et al. Periocular and orbital amyloidosis: clinical characteristics, management, and outcome. Ophthalmology 2006; 113: 1657-1664
- 217 Mora-Horna ER, Rojas-Padilla R, Lopez VG. et al. Ocular adnexal and orbital amyloidosis: a case series and literature review. Int Ophthalmol 2016; 36: 281-298
- 218 Naxer S, Behnes CL, Schittkowski MP. [Amyloidosis--a rare differential diagnosis of an orbital tumour]. Klin Monbl Augenheilkd 2011; 228: 555-564
- 219 Ladner-Merz S, Muller-Ladner U. [Amyloidoses]. Z Rheumatol 2008; 67: 677-682 quiz 683
- 220 Khaira M, Mutamba A, Meligonis G. et al. The use of radiotherapy for the treatment of localized orbital amyloidosis. Orbit 2008; 27: 432-437
- 221 Eghbali A, Hassan S, Seehra G. et al. Ophthalmological findings in Gaucher disease. Mol Genet Metab 2019; 127: 23-27
- 222 Prakalapakorn SG, Proia AD, Yanovitch TL. et al. Ocular and histologic findings in a series of children with infantile pompe disease treated with enzyme replacement therapy. J Pediatr Ophthalmol Strabismus 2014; 51: 355-362
- 223 Slingerland NW, Polling JR, van Gelder CM. et al. Ptosis, extraocular motility disorder, and myopia as features of pompe disease. Orbit 2011; 30: 111-113
- 224 Winter AW, Salimi A, Ospina LH. et al. Ophthalmic manifestations of Gaucher disease: the most common lysosomal storage disorder. Br J Ophthalmol 2019; 103: 315-326
- 225 Yanovitch TL, Banugaria SG, Proia AD. et al. Clinical and histologic ocular findings in pompe disease. J Pediatr Ophthalmol Strabismus 2010; 47: 34-40
- 226 Wabbels B, Ali N, Kunz WS. et al. [Chronic progressive external ophthalmoplegia and Kearns-Sayre syndrome : interdisciplinary diagnosis and therapy]. Ophthalmologe 2008; 105: 550-556
- 227 Pfeiffer MJ. [Chronic Progressive External Ophthalmoplegia Ptosis: Problems with Diagnostics and Treatment]. Klin Monbl Augenheilkd 2018; 235: 31-33
- 228 Pitceathly RD, Morrow JM, Sinclair CD. et al. Extra-ocular muscle MRI in genetically-defined mitochondrial disease. Eur Radiol 2016; 26: 130-137
- 229 Rahman S, Blok RB, Dahl HH. et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996; 39: 343-351
- 230 McKelvie P, Infeld B, Marotta R. et al. Late-adult onset Leigh syndrome. J Clin Neurosci 2012; 19: 195-202
- 231 Lake NJ, Compton AG, Rahman S. et al. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol 2016; 79: 190-203
- 232 Han J, Lee YM, Kim SM. et al. Ophthalmological manifestations in patients with Leigh syndrome. Br J Ophthalmol 2015; 99: 528-535
- 233 Fichter N, Schittkowski M, Guthoff R. Erkrankungen der Tränendrüse. Ophthalmologe 2005; 102: 399-425
- 234 Moreiras J. Tumors of the lacrimal gland - Orbit – examination, diagnosis, mircrosurgery, pathology. 2004
- 235 Font R, Gamel J. Epithelial tumours of the lacrimal gland: an analysis of 265 cases.In Jakobiec F (ed) Ocular and adnexal tumours. Aesculapius, Birmingham. 1978
- 236 Riedel K, Markl A, Hasenfratz G. et al. Epithelial tumors of the lacrimal gland: Clinico-pathologic correlation and management. Neurosurg Rev 1990; 13: 289-298
- 237 Andreasen S, Esmaeli B, Holstein L. et al. An Update on Tumors of the Lacrimal Gland. Asia-Pac. J Ophthalmol 2017; 6: 159-172
- 238 Cates C, Manners R, Rose G. Pleomorphic adenoma of the lacrimal gland in a 10 year old girl. Br J Ophthalmol 2002; 86: 249-250
- 239 Faktorovich E, Crawford J, Char D. Benign mixed tumor (pleomorphic adenoma) of the lacrimal gland in a 6-year-old boy. Am J Ophthalmol 1996; 122: 446-447
- 240 Mercado G, Gunduz K, Shields C. et al. Pleomorphic adenoma of the lacrimal gland in a teenager. Arch Ophthalmol 1998; 116: 962-963
- 241 Ni C, Kuo P, Dryja T. Histopathological classification of 272 primary epithelial tumors of the lacrimal gland. Chin Med J 1992; 105: 481-485
- 242 Rose G, Wright J. Pleomorphic adenoma of the lacrimal gland. Br J Ophthalmol 1992; 76: 395-400
- 243 Stefko S, DiBernardo C, Green W. et al. Pleomorphic adenoma of the lacrimal gland with extensive calcification. Arch Ophthalmol 2004; 122: 778-780
- 244 Stupp T, Pavlidis M, Buchner T. Pleomorphic Adenoma of the Lacrimal Gland in a Child After Treatmentof Acute Lymphoblastic Leukemia. Arch Ophthalmol 2004; 122: 1538-1540
- 245 Bonavolontà G, Tranfa F, Staibano S. et al. Warthin Tumor of the Lacrimal Gland. Am J Ophthalmol 1997; 124: 857-858
- 246 Ghosh D, Ekka J. Adenolymphoma of the lacrimal gland: A very rare case report. Int J Ocul Oncol oculoplasty 2018; 4: 144-145
- 247 Bartoletti-Stella A, Salfi N, Ceccarelli C. et al. Mitochondrial DNA Mutations in Oncocytic Adnexal Lacrimal Glands of the Conjunctiva. ARCH OPHTHALMOL 2011; 129: 664-666
- 248 Calle C, Castillo I, Eagle R. et al. Oncocytoma of the Lacrimal Gland: Case Report and Review of the Literature. Orbit 2006; 25: 243-247
- 249 Cameron J, Barras C. Oncocytoma of the eyelid. ACTA OPHTHALMOL SCAND 2004; 83: 125
- 250 George E, Swanson P, Newman B. et al. Oculocutaneous oncocytic tumors: clinicopathologic and immunohistochemical study of 2 cases with literature review. Am J Dermatopathol 2007; 29: 279-285
- 251 Morand B, Bettega G, Bland V. et al. Oncocytoma of the eyelid: an aggressive benign tumor. Ophthalmology 1998; 105: 2220-2224
- 252 Shields C, Shields J. Tumors of the conjunctiva and cornea. Survey of Ophthalmology 2004; 49: 3-24
- 253 Shields C, Shields J, Arbizo V. Oncocytoma of the caruncle. Am J Ophthalmol 1986; 102: 315-319
- 254 Surakiatchanukul T, Sioufi K, Pointdujour-Lim R. et al. Caruncular Oncocytoma Mimicking Malignant Melanoma. Ocul Oncol Pathol 3: 320-323
- 255 Bolzoni A, Pianta L, Farina D. et al. Benign myoepithelioma of the lacrimal gland: report of a case. Eur Arch Otorhinolaryngol 2005; 262: 186-188
- 256 Font R, Garner A. Myoepithelioma of the lacrimal gland: report of a case with spindle cell morphology. Brit J Ophthalmol 1992; 76: 634-636
- 257 Grossniklaus H, Wojno T, Wilson M. et al. Myoepithelioma of the Lacrimal Gland. Arch Ophthalmol 1997; 115: 1588-1590
- 258 Heathcote J, Hurwitz J, Dardick I. A Spindle-Cell Myoepithelioma of the Lacrimal Gland. Arch Ophthalmol 1990; 108: 1135-1139
- 259 Nickol T, E R Kleinsasser O. Myoepitheliome der Parotis. Jahresversammlung der Deutschen Gesellschaft für HNO-Heilkunde. 1998
- 260 Bajaj M, Pushker N, Kashyap S. Cystadenoma of the lacrimal gland. Orbit 2002; 21: 301-305
- 261 Bernardini F, Devoto M. JO C. Epithelial tumors of the lacimal gland: an update. Curr Opin in Ophthalmol 2008; 19: 409-413
- 262 Silbert J, Braich P, Misra R. Basal cell cystadenoma of the lacrimal gland: diagnostic pitfalls of a basaloid pattern in lacrimal tumours. Internat Ophthalmol. 2011 DOI: 43-46
- 263 Wang L, Zhang SK, Ma Y. et al. Papillary cystadenoma of the parotid gland: A case report. World J Clin Cases 2019; 7: 366-372
- 264 Pfeiffer M, Yin V, Bell D. et al. Sclerosing Polycystic Adenosis of the Lacrimal Gland. Ophthalmology 2013; 120: 873
- 265 Speight P, Barrett A. Salivary gland tumours: diagnostic challenges and an update on the latest WHO classification. Diagn Histopathol 2020; 26: 147-158
- 266 Brunnmann R, Ro J, Ordonez N. et al. Extrapleural Solitary Fibrous Tumor: A Clinicopathologic Study of 24 Cases. Mod Pathol 1999; 12: 1034-1042
- 267 Mupas-Uy J, Kitaguchia Y, Takahashia Y. et al. Solitary Fibrous Tumor in the Lacrimal Gland Fossa: A Case Report. Case Rep Ophthalmol 2016; 7: 398-403
- 268 Okike N, Bernatz P, Woolner L. Localized mesothelioma of the pleura. Benign and malignant variants. J Thorac Cardiovasc Surg 1978; 75: 363-372
- 269 Scott I, Tanenbaum M, Rubin D. Solitary fibrous tumor of the lacrimal gland fossa. Ophthalmology 1996; 103: 1613-1618
- 270 Vallat-Decouvelaere A, Dry S, Fletcher C. Atypical and malignant solitary fibrous tumor in extrathoracic locations; evidence of their comparability to intra-thoracic tumors. Am J Surg Pathol 1998; 22: 1501-1511
- 271 Woo K, Suh Y, Kim Y. Solitary fibrous tumor of the lacrimal sac. Ophthalmic Plast Reconstr Surg 1999; 15: 450-453
- 272 Bielory BP, Turbin RE, Mirani NM. et al. Radiographic and histological findings in an atypical orbital myxoma. Ophthalmol Eye Dis 2011; 3: 55
- 273 Demirci H, Shields CL, Eagle RCJ. et al. Report of a conjunctival myxoma case and review of the literature. Arch Ophthalmol 2006; 124: 735-738
- 274 Kini Rao A, Nayal B. Conjunctival myxoma-a case report. Malays J Med Sci 2013; 20: 92-94
- 275 Parikh D, Mukherjee B. Lacrimal gland myxoma. Indian J Ophthalmol 2017; 65: 887-889
- 276 Rambhatla S, Subramanian N, Gangadhara Sundar JK. et al. Myxoma of the orbit. Indian J Ophthalmol 2003; 51: 85-87
- 277 Bajaj M, Pushker N, Fibrous SK. histiocytoma of the lacrimal gland. Ophthal Plast Reconstr Surg 2007; 23: 145-147
- 278 Andrew N, Coupland S, Pirbhai A. et al. Lymphoid hyperplasia of the orbit and ocular adnexa: A clinical pathologic review. Survey of Ophthalmology 2016; 61: 778-790
- 279 Shields J, Shields C, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions: the 2002 Montgomery lecture, part 1. Ophtalmology. 2004 DOI: 997-1008
- 280 Wittekind C. TNM-Klassifikation maligner Tumoren. Achte Auflage. Aufl. WILEY-VCH; 2017
- 281 Devoto M, Croxatto J. Primary cystadenocarcinoma of the lacrimal gland. Ophthalmology 2003; 110: 2006-2010
- 282 Takahira M, Nakamura Y, Shimizu S. et al. Carcinosarcoma of the Lacrimal Gland Arising From a Pleomorphic Adenoma. Am J Ophthalmol 2005; 140: 340-341
- 283 Daisuke Y, Toshiyuki O, Shuichi Y. Case of primary neuroendocrine carcinoma in lacrimal gland. Acta Ophthalmol 2013; 91: 252
- 284 Gess A, Silkiss R. A merkel cell carcinoma of the lacrimal gland. Ophthal Plast Reconstr Surg. 28. 11-13
- 285 El-Naggar A, Chan J, Grandis J. World Health organization classification of head and neck tumours. 2017 DOI:
- 286 Weis E, Rootman J, Joly T. et al. Epithelial lacrimal gland tumors: pathologic classification and current understanding. Arch Ophthalmol 2009; 127: 1016-1028
- 287 Gamel J, Font R. Adenoidcystic carcinoma of the lacrimal gland: the clinical significance of a basaoid histologic pattern. Human Pathol 1982; 13: 219-225
- 288 Friedrich R, Bleckmann V. Adenoid cystic carcinoma of the salivary and lacrimal glands origin: localization, classification, clinical pathological correlation, treatment results and long-term follow-up control in 84 patients. Anticancer Res 2003; 23: 937-940
- 289 Goldberg R. Intraarterial chemotherapy. A welcome new idea for the management of adenoidcystic carcinoma of the lacrimal gland. Arch Ophthalmol 1998; 116: 372-373
- 290 Hamper K, Lazar F, Dietel M. Prognostic factors for adenoid cystic carcinoma of the head and neck: a retrospectiv evaluation of 96 cases. J Oral Pathol Med 1990; 19: 101-107
- 291 Henderson J. In Orbital Tumors New York: Raven Press;. 1994: 323-342
- 292 Lee D, Campbell R, Waller R. A clinicopathologic study of primary adenoid cystic carcinoma of the lacrimal gland. Ophthalmology 1985; 92: 128-134
- 293 Strianese D, Baldi G, Staibano S. Expression of apoptosis-related markers in malignant epithelial tumors of the lacrimal gland and their relation to clinical outcome. Br J Ophthalmol 2007; 91: 1239-1243
- 294 Tse D, Benedetto P, Dubovy S. Clinical analysis oft he effect on intraarterial cytoreductive chemotherapy in the treatment of lacrimal gland andoid cystic carcinoma. Am J Ophthalmol 2006; 141: 44-53
- 295 Wright J, Rose G, Garner A. Primary malignant neoplasms of the lacrimal gland. Br J Ophthalmol 1992; 76: 401-407
- 296 Karcioglu Z. Orbital Tumors. Diagnosis and Management. New York: Springer; 2005
- 297 Ahmad S, Esmaeli B, Williams M. American Joint Committee on Cancer classification predicts outcome of patients with lacrimal gland adenoid cystic carcinoma. Ophthalmology 2009; 116: 1210-1215
- 298 GROUP IHANS. Cervical lymph node metastasis in adenoid cystic carcinoma of the sinonasal tract, nasopharynx, lacrimal glands and external auditory canal: a collective international review. J Laryngol Otol 2016; 130: 1093-1097
- 299 Xiao R, Sethi R, Feng A. et al. The role of elective neck dissection in patients with adenoid cystic carcinoma of the head and neck. Laryngoscope 2019; 129: 2094-2104
- 300 Bördlein I. Mit schweren Ionen gegen hartnäckige Tumoren. Deutsches Ärzteblatt 2004; 101: 1718-1720
- 301 Hayashi K, Koto M, Ikawa H. et al. Efficacy and safety of carbon-ion radiotherapy for lacrimal gland carcinomas with extraorbital extension: a retrospective cohort study. Oncotarget 2018; 9: 12932-12940
- 302 Lesueur P, Rapeaud E, De Marzi L. et al. Adenoid Cystic Carcinoma of the Lacrimal Gland: High Dose Adjuvant Proton Therapy to Improve Patients Outcomes. Front Oncol 2020; 10: 135
- 303 Wolkow N, Jakobiec F, Lee H. et al. Long-term Outcomes of Globe-Preserving Surgery With Proton Beam Radiation for Adenoid Cystic Carcinoma of the Lacrimal Gland. Am J Ophthalmol. 2018 195.
- 304 Shields J, Shields C, Freire J. Plaque radiotherapy for selected orbital malignancies: preliminary observations: hte 2002 Montgomery Lecture, part 2. Ophthal Plast Reconstr Surg 2003; 19: 91-95
- 305 Tyl J, Blank L, Koornneef L. Brachytherapy in orbital tumors. Ophthalmology 1997; 104: 1475-1479
- 306 Gündüz A, Yesiltas Y, Shileds C. Overview of benign and malignant lacrimal gland tumors. Curr Opin Ophthalmol 2018; 29: 458-468
- 307 Ashton N. Epithelial tumors of the lacrimal gland. Mod Probl Ophthalmol 1975; 14: 306-323
- 308 Perzin K, Jakobiec F, Lovolsi V. Lacrimal gland malignant mixed tumors (carcinomas arising in benign mixed tumors): a clinicopathologic study. Cancer 1980; 45: 2593-2606
- 309 Shields C, Shields J, Eagle R. Clinicopathologic review of 142 cases of lacrimal gland lesions. Ophthalmology 1989; 96: 431-435
- 310 Henderson J, Farrow G. Primary malignant mixed tumors of the lacrimal gland. Report of 10 cases. Ophthalmology 1980; 87: 466-475
- 311 Jakobiec F, Bilyk J, Font R. Ophthalmic Pathology. An Atlas and Textbook. 4th ed. Aufl. Philadelphia. WB Saunders; 1996
- 312 Chen A, Garcia J, Bucci M. et al. The role of postoperative radiation therapy in carcinoma ex pleomorphic adenoma of the parotid gland. Int J Radiat Oncol Biol Phys 2007; 67: 138-143
- 313 Takahira M, Minato H, Takahashi M. Cystic carcinoma ex pleomorphic adenoma of the lacrimal gland. Ophthal Plast Reconstr Surg 2007; 23: 407-409
- 314 Hwang S, Kim K. High-grade Mucoepidermoid Carci- noma of the Lacrimal Gland. Korean J Ophthalmol 2018; 32: 426-427
- 315 Heaps R, Miller N, Albert D. Primary adenocarcinoma of the lacrimal gland. Ophtalmology 1993; 100: 1856-1860
- 316 von Holstein S, Coupland S. , D B. Epithelial Tumors of the Lacrimal Gland: a clinical, histopathological, surgical, and oncological survey. Acta Ophthalmol 2013; 91: 195-206
- 317 Dennie T. Metastastic her-2 amplified lacrimal gland carcinoma with response to lapatinib treatment. Case Rep Oncol Med 2015; 2015: 1-3
- 318 Totuk O, Demir M, Yapicier O. et al. Low-Grade Mucoepidermoid Carcinoma of the Lacrimal Gland in a Teenaged Patient. Case Rep Ophthalmol Med. 2017
- 319 Eviatar J, Hornblass A. Mucoepidermoid carcinoma of the lacrimal gland: 25 cases and a review and update of the literature. Ophthal Plast Reconstr Surg 1993; 9: 170-181
- 320 Sofinski S, Brown B, Rao N. Mucoepidermoid carcinoma of the lacrimal gland. Case report and review of the literature. Ophthalmic Plast Reconstr Surg 1986; 2: 147-151
- 321 Thorvaldsson S, Beahrs O, Woolner L. et al. Mucoepidermoid tumors of the major salivary glands. Am J Surg 1970; 120: 432-438
- 322 Katz E, Rootman J, Dolman P. et al. Primary Ductal Adenocarcinoma of the Lacrimal Gland. Ophthalmology 1996; 103: 157-162
- 323 Yang H, Wu C, Tsai C. et al. Primary ductal adenocarcinoma of lacrimal gland: Two case reports and review of the literature. Taiwan J Ophthalmol 2018; 8: 42-48
- 324 Ishida M, Hotta M, Kushima R. et al. Case of ductal adenocarcinoma ex pleomorphic adenoma of the lacrimal gland. Rinsho Byori 2009; 57: 746-751
- 325 Andreasen S, Grauslund M, Heegaard S. Lacrimal gland ductal carcinomas: clinical, morphological and genetic characterization and implications for targeted treatment. Acta Ophthalmol 2017; 95: 299-306
- 326 Jaspers H, Verbist B, Schoffelen R. et al. Androgen Receptor–Positive Salivary Duct Carcinoma: A Disease Entity With Promising New Treatment Options. J Clin Oncol 2011; 29: 473-476
- 327 See T, Stålhammar G, Tang T. et al. Primary ductal adenocarcinoma of the lacrimal gland: A review and report of five cases. Survey of Ophthalmology 2020; 65: 371-380
- 328 Anesi A, Negrello S, Lucchetti D. et al. Clinical Management of Acinic Cell Carcinoma of the Lacrimal Gland. Case Rep Oncol 2019; 12: 777-790
- 329 De Rosa G, Zeppa P, Tranta F. Acinic Cell Carcinoma in a Lacrimal Gland: first case report. Cancer 1986; 57: 1988-1991
- 330 Jang J, Kie J, Lee S. Acinic cell carcinoma of the lacrimal gland with intracranial extension. Ophthalmic Plast Reconstr Surg 2001; 17: 454-457
- 331 Rosenbaum P, Mahadevia P, Goodman L. Acinic cell carcinoma of the lacrimal gland. Arch Ophthalmol 1995; 113: 781-785
- 332 Briscoe D, Mahmood S, Bonshek R. et al. Primary sebaceous carcinoma of the lacrimal gland. Br J Ophthalmol 2001; 85: 625-626
- 333 Harvey P, Parsons M, Rennie I. Primary sebaceous carcinoma of lacrimal gland: a previously unreported primary neoplasm. Eye 1994; 8: 592-595
- 334 Kiratli H, Tarlan B, Fırat P. Primary sebaceous carcinoma of the lacrimal gland. Orbit 2012; 31: 352-354
- 335 Park H, Choi S. Primary sebaceous carcinoma of lacrimal gland: A case report and review of literature. World J Clin Cases 2018; 6: 1194-1198
- 336 Yamamoto N, Mizoe J, Hasegawa A. et al. Primary sebaceous carcinoma of the lacrimal gland treated by carbon ion radiotherapy. Int J Clin Oncol 2003; 8: 386-390
- 337 Herrera G. Light microscopic, ultrastructural and immunocytochemical spectrum of malignant lacrimal and salsivary gland tumors, including malignant mixed tumors. Pathobiology 1990; 58: 312-322
- 338 Iida K, Shikishima K, Okido M. et al. A case of malignant myoepithelioma in the lacrimal gland. Nippon Ganka Gakkai Zasshi 2001; 105: 42-46
- 339 Larbcharoensub N, Pangpunyakulchai D, Aroonroch R. et al. Lacrimal myoepithelial carcinoma ex recurrent pleomorphic adenoma: A clinicopathological report and review of the literature. Mol Clin Oncol 2018; 8: 209-213
- 340 Nagao T, Sugano I, Ishida Y. Salivary gland mmalignant myoepithelioma: a clinicopathologic and immunohistochemical study of ten cases. Cancer 1998; 83: 1292-1299
- 341 Xu T, Liao Z, Tang J. et al. Myoepithelial carcinoma of the headand neck: A report of 23 casesand literature review. Cancer Treatment. Communications 2014; 2: 24-29
- 342 Mahdi Y, Azami M, Daoudi R. et al. Diagnostic pitfall: primary myoepithelial carcinoma of the lacrimal gland, case report and literature review. BMC Clinical Pathology 2018; 18: 1-7
- 343 Yu G, Ma D, Sun K. Myoepithelial carcinoma of the salivary glands: behavior and management. Chin Med J 2003; 116: 163-165
- 344 Soumarová R, Lovasová Z, Vaňková M. Radiotherapy in malignant myoepithelioma of the soft palate – case report. Case Rep Oncol 2009; 2: 116-120
- 345 Nieder C, Schneller F, Grosu A. Radiotherapy and chemotherapy for myoepithelioma of the sellar region. Strahlenther Onkol 2005; 181: 260-263
- 346 Fenton S, Srinivasan S, Harnett A. et al. Primary squamous cell carcinoma of the lacrimal gland. Eye 2003; 17: 424-425
- 347 Su G, Patipa M, Font R. Primary squamous cell carcinoma arising from an epithelium-lined cyst of the lacrimal gland. Ophthal Plast Reconstr Surg 2005; 21: 383-385
- 348 Hotta K, Arisawa T, Mito H. et al. Primary squamous cell carcinoma of the lacrimal gland. Clin Experiment Ophthalmol 2005; 33: 534-536
- 349 Bernardini F, Orcioni G, Croxatto J. Oncocytic carcinoma of the lacrimal gland in a patient with neurofibromatosis. Ophthal Plast Reconstr Surg 2010; 26: 486-488
- 350 Kalantzis G, Norris J, El-Hindy N. et al. Oncocytic adenocarcinoma of the lacrimal gland: an unusual presentation. Eye 2013; 27: 104-105
- 351 Selva D, Davis G, Dodd T. Polymorphous Low-Grade Adenocarcinoma of the Lacrimal Gland. ARCH OPHTHALMOL 2004; 122: 915-917
- 352 Bortz J, Zhang P, Eagle R. et al. Secretory Carcinoma of the Lacrimal Gland: A Rare Case Report. Ophthalmic Plast Reconstr Surg 2018; 34: 154-157
- 353 Hyrczaa M, Andreasen S, Melchiorc L. et al. Primary Secretory Carcinoma of the Lacrimal Gland: Report of a New Entity. Am J Ophthalmol 2018; 193: 178-183
- 354 Kadir S. Synovial Sarcoma involving lacrimal gland: A rare clinical entity. Int J Ocu Oncol Oculoplast. 1: 14–18
- 355 Likhvantseva V, Safonova T, Kuzmin K. Primary granulocytic sarcoma of lacrimal gland. Vestn Oftalmol 2016; 132: 82-89
- 356 Yilmaz A, Saydam G, Sahin F. et al. Granulocytic sarcoma: a systematic review. Am J Blood Res 2013; 3: 265-270
- 357 Rajabi M, Riazi H, Hosseini S. et al. Malignant Peripheral Nerve Sheath Tumor of Lacrimal Nerve: A Case Report. Orbit 2015; 34: 41-44
- 358 Stark A, Hugo H, Buhl R. et al. Tumoren peripherer Nerven. Dtsch Arztebl 2002; 99: 928-933
- 359 Gündüz K, Shields J, Eagle R.. Malignant Rhabdoid Tumor of the Orbit. Arch Ophthalmol 1998; 116: 243-246
- 360 Johnson L, Sexton F, Goldberg S. Poorly differentiated primary orbital sarcoma (presumed malignant rhabdoid tumor): radiologic and histopathologic correlation. Arch Ophthalmol 1991; 109: 1275-1278
- 361 Orphanet. Rhabdoidtumor. In: Orphanet; 2020
- 362 Rootman J, Damji K, Dimmick J. Malignant rhabdoid tumor of the orbit. Ophthalmology 1989; 96: 1650-1654
- 363 Walford N, Deferrai R, Slater R. Intraorbital rhabdoid tumour following bilateral retinoblastoma. Histopathology 1992; 20: 170-173
- 364 Niffenegger J, Jakobiec F, Shore J. et al. Adult extrarenal rhabdoid tumor of the lacrimal gland. Ophthalmology 1992; 99: 567-574
- 365 Ferry J, Fung C, Zukerberg L. et al. Lymphoma of the ocular adnexa: A study of 353 cases 2007; 31: 170-184
- 366 Rasmussen P, Ralfkiaer E, Prause J. Malignant lymphoma of the lacrimal gland. A nation-based study. Arch Ophthalmol 2011; 129: 1275-1280
- 367 Johnson T, Tse D, Byrne G. Ocular adnexal lymphoid tumors: a clinicopathologic and molecular genetic study of 77 patients. Ophthal Plast Reconstr Surg 1999; 15: 171-179
- 368 Polito E, Galieni P, Leccisotti A. Clinical and radiological presentation of 95 orbital lymphoid tumors. Graefes Arch Clin Exp Ophthalmol 1996; 234: 504-509
- 369 Aranow M. Ocular adnexal lymphoma: evidence-based treatment approach. Int Ophthalmol Clin 2015; 55: 97-109
- 370 König L, Stade R, Rieber J. Radiotherapy of indolent orbital lymphomas: two radiation concepts. Strahlenther Onkol 2016; 192: 414-421
- 371 de Leval L, Jaffe E. Lymphoma Classification. Cancer J 2020; 26: 176-185
- 372 Shields J, Shields C, Eagle R. Metastatic renal cell carcinoma to the palpebral lobe of the lacrimal gland. Ophthal Plast Reconstr Surg 2001; 17: 191-194
- 373 Fowler J, Terplan K. Fibroma of the Orbit. Arch Ophthalmol 1942; 28: 263-271
- 374 Mortada A. Fibroma of the orbit. Brit J Ophthal 1971; 55: 350
- 375 Rong A, Ulloa-Padilla J, Blessing N. et al. Subperiosteal fibroma of the orbit. Orbit 2018; 37: 378-380
- 376 Rabelo N, da Silva V, do Espírito Santo M. et al. Orbit ossifying fibroma – Case report and literature review. Surg Neurol Int 2020; 11: 35
- 377 Sheikh BY. Cranial aneurysmal bone cyst “with special emphasis on endovascular management”. Acta Neurochir (Wien) 1999; 141: 601-610 discussion 610-601
- 378 Glien A, Meyer B, Wernert N. et al. [Orbital aneurysmal bone cyst in a 2-year-old child]. HNO 2007; 55: 281-286
- 379 Wang JC, Zhang M, Zhao XX. Aneurysmal bone cyst of the orbit. Chin Med J (Engl) 2015; 128: 562-563
- 380 Yu JW, Kim KU, Kim SJ. et al. Aneurysmal bone cyst of the orbit : a case report with literature review. J Korean Neurosurg Soc 2012; 51: 113-116
- 381 Ruiter DJ, van Rijssel TG, van der Velde EA. Aneurysmal bone cysts: a clinicopathological study of 105 cases. Cancer 1977; 39: 2231-2239
- 382 Biesecker JL, Marcove RC, Huvos AG. et al. Aneurysmal bone cysts. A clinicopathologic study of 66 cases. Cancer 1970; 26: 615-625
- 383 Schajowicz F. Tumors and tumorlike lesions of bone 2. Aufl. Berlin-Heidelberg-New York: Springer-Verlag; 1994
- 384 Oliveira AM, Chou MM. USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum Pathol 2014; 45: 1-11
- 385 Fay A, Dolman PJ. Laboratory Serologic Investigations.In Fay A, Dolman PJ , Hrsg. Diseases and Disorders of the Orbit and Ocular Adnexa E-book. Elsevier; 2017: 314
- 386 Mallen JR, Caranfa JT, Zimmerman D. et al. Atypical presentation of a solid-variant orbital aneurysmal bone cyst with a literature review. Neurochirurgie 2018; 64: 431-433
- 387 Chawla B, Khurana S, Kashyap S. Giant cell reparative granuloma of the orbit. Ophthalmic Plast Reconstr Surg 2013; 29: e94-95
- 388 Kamoshima Y, Sawamura Y, Imai T. et al. Giant cell tumor of the frontal bone in a girl: case report. Neurol Med Chir (Tokyo) 2011; 51: 798-800
- 389 Smolarz-Wojnowska A, Essig H, Gellrich NC. et al. [Orbital tumours in children and adolescents. Differential diagnostics and clinical symptoms]. Ophthalmologe 2010; 107: 543-548
- 390 Vernet O, Ducrey N, Deruaz JP. et al. Giant cell tumor of the orbit. Neurosurgery 1993; 32: 848-851
- 391 Yip CM, Lee HP, Hsu SS. et al. Left orbital roof giant cell tumor of bone: A case report. Surg Neurol Int 2018; 9: 127
- 392 Yamaguchi T, Dorfman HD. Giant cell reparative granuloma: a comparative clinicopathologic study of lesions in gnathic and extragnathic sites. Int J Surg Pathol 2001; 9: 189-200
- 393 de Lange J, van den Akker HP, van den Berg H. Central giant cell granuloma of the jaw: a review of the literature with emphasis on therapy options. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007; 104: 603-615
- 394 Auclair PL, Cuenin P, Kratochvil FJ. et al. A clinical and histomorphologic comparison of the central giant cell granuloma and the giant cell tumor. Oral Surg Oral Med Oral Pathol 1988; 66: 197-208
- 395 de Lange J, van Rijn RR, van den Berg H. et al. Regression of central giant cell granuloma by a combination of imatinib and interferon: a case report. Br J Oral Maxillofac Surg 2009; 47: 59-61
- 396 Roman-Romero L, Gonzalez-Garcia R. Cholesterol granuloma of the orbit. Report of cases and analysis of controversial treatment. J Maxillofac Oral Surg 2011; 10: 166-169
- 397 Loeffler KU, Kommerell G. Cholesterol granuloma of the orbit--pathogenesis and surgical management. Int Ophthalmol 1997; 21: 93-98
- 398 Selva D, Phipps SE, O'Connell JX. et al. Pathogenesis of orbital cholesterol granuloma. Clin Exp Ophthalmol 2003; 31: 78-82
- 399 Daus W, Voges J, Schwechheimer K. et al. [Cholesterol granuloma of the orbits--clinicopathologic study]. Klin Monbl Augenheilkd 1988; 193: 195-199
- 400 Hsu HT, Liao WC, Wu CC. et al. Cholesterol granuloma of the orbit. Kaohsiung J Med Sci 2017; 33: 422-423
- 401 Rong AJ, Erickson BP, Blessing NW. et al. Orbital cholesterol granuloma: A report and discussion of orbital findings. Am J Ophthalmol Case Rep 2019; 15: 100468
- 402 Arat YO, Chaudhry IA, Boniuk M. Orbitofrontal cholesterol granuloma: distinct diagnostic features and management. Ophthalmic Plast Reconstr Surg 2003; 19: 382-387
- 403 Aferzon M, Millman B, O'Donnell TR. et al. Cholesterol granuloma of the frontal bone. Otolaryngol Head Neck Surg 2002; 127: 578-581
- 404 Kabra R, Patel S, Shanbhag S. Orbital Chondroma: A rare mesenchymal tumor of orbit. Indian J Ophthalmol 2015; 63: 551-554
- 405 Garrity J, Henderson J. Henderson’s Orbital Tumors.In 4th ed. Aufl. USA: Lippincott Williams and Wilkis; 2007: 103–104
- 406 Rootman J, Connell J. Primary bone tumors of orbit. Surv Ophthalmol 2004; 49: 328-342
- 407 Shields J, Bakewell B, Augsburger J. et al. Classification and incidence of space-occupying lesions of the orbit. A survey of 645 biopsies. Arch Ophthalmol 1984; 102: 1606-1611
- 408 Pushker N, Meel R, Sharma S. et al. Giant orbital Schwannoma with fluid-fluid-levels. Br J Ophthalmol 2010; 95: 1168
- 409 Misra S, Gogri P, Misra N. et al. Recurrent neurofibroma of the orbit. Australas Med J 2013; 30: 189-191
- 410 Lee L, Gigantelli J, Kincaid M. Localized neurofibroma of the orbit: a radiographic and histopathologic study. Ophthal Plast Reconstr Surg 2000; 16: 241-246
- 411 Turbin R, Thompson C, Kennerdell J. et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophtalmology 2002; 109: 890-899
- 412 Schittkowski M, Fichter N., Orbitale Neoplasien Teil I. Einführung, vaskuläre und neurogene Neoplasien. Ophthalmologe 2012; 109: 1033-1046
- 413 Jain D, Ebrahimi K, Miller N. et al. Intraorbital meningiomas: a pathologic review using current World Health Organization criteria. Arch Pathol Lab Med 2010; 134: 766-770
- 414 Eddleman C, Liu J. Optic nerve sheath meningioma: current diagnosis and treatment. Neurosurg Focus 2007; 23: E4
- 415 Saeed P, Rootman J, Nugent R. et al. Optic nerve sheath meningiomas. Ophthalmology 2003; 110: 2019-2030
- 416 Ramey W, Arnold S, Chiu A. et al. A Rare Case of Optic Nerve Schwannoma: Case Report and Review of the Literature. Cureus 2015; 7: e265
- 417 Gnekow A. Low-grade chiasmatic-hypothalamic glioma-carboplatin and vincristine chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy - report from the multicenter treatment study for children and adolescents with a low grade glimoa - HIT-LGG 1996 - of the Society of Paediatric Oncology and Hematology (GPOH). Klin Padiatr 2004; 216: 331-342
- 418 Sharif S. Second primary tumors in neurofibromatosis 1 patients treated for optic nerve glioma: substantial risks after radiotherapy. J Clin Oncol 2006; 24: 2570-2575
- 419 Wilhelm H. Primary optic nerve tumors. Curr Opin Neurol 2009; 22: 11-18
- 420 Emesz M, Arlt E, Krall E. et al. Granularzelltumor der Orbita. Der. Ophthalmologe 2014; 111: 866-870
- 421 Huang N, Rayess H, Svider P. et al. Orbital Paraganglioma: A Systematic Review. J Neurol Surg B Skull Base 2017; 79: 407-412
- 422 Makhdoomi R, Nayil K, Santosh V. et al. Orbital paraganglioma--a case report and review of the literature. Clin Neuropathol 2010; 29: 100-104
- 423 Al-Khiary H, Ayoubi A, Elkhamary S. Primary orbital ganglioneuroma in a 2-year-old healthy boy. Saudi J Ophthalmol 2010; 24: 101-104
- 424 Cannon T, Brown H, Hughes B. Orbital Ganglioneuroma in a Patient With Chronic Progressive Proptosis. Arch Ophthalmol 2004; 122: 1712-1714
- 425 Hatsukawa Y, Furukawa A, Kawamura H. et al. Rhabdomyoma of the orbit in a child. Am J Ophthalmol 1997; 123: 142-144
- 426 Li Y, Li N, Zang W. et al. Rhabdomyoma of the orbit. J Pediatr Ophthalmol Strabismus 2008; 45: 113-115
- 427 Myung J, Kim I, Chun J et al. Rhabdomyoma of the orbit: a case report. Pediatric Radiolog. y. 32: 589–592
- 428 Gündüz K, Günalp I, Erden E. et al. Orbital leiomyoma: report of a case and review of the literature. Surv Ophthalmol 2004; 49: 237-242
- 429 Merani R, Khannah G, Mann S. et al. Orbital leiomyoma: a case report with clinical, radiological and pathological correlation. Clin Exp Ophthalmol 2005; 33: 408-411
- 430 Shah N, Chang W, White V. et al. Orbital lipoma: 2 cases and review of literature. Ophthalmic Plast Reconstr Surg 2007; 23: 202-205
- 431 Franchi G, Sleilati E, Soupre V. et al. Melanotic neuroectodermal tumour of infancy involving the orbit and maxilla: surgical management and follow-up strategy. Br J Plast Surg 2002; 55: 526-529
- 432 Koob M, Fayard C, Pariente D. et al. Prenatal diagnosis of orbital melanotic neuroectodermal tumor in infancy. Ultrasound Obstet Gynecol 2015; 46: 247-252
- 433 Soles B, Wilson A, Lucas D. et al. Melanotic Neuroectodermal Tumor of Infancy. Arch Pathol Lab Med 2018; 142: 1358-1363
- 434 NIH. Childhood Rhabdomyosarcoma Treatment (PDQ®)–Health Professional Version. In: National Cancer Institute at the National Institutes of Health; 2020
- 435 Al-Bargi H, Al-Maiman M. Orbital fibrosarcoma: a case report and literature review. Egypt J Oral Maxillofac Surg 2012; 3: 45-48
- 436 Bahrami A, Folpe A. Adult-type fibrosarcoma: A reevaluation of 163 putative cases diagnosed at a single institution over a 48-year period. Am J Surg Pathol 2010; 34: 1504-1513
- 437 Scruggs B, Ho S, Valenzuela A. Diagnostic challenges in primary orbital fibrosarcoma: a case report. Clin Ophthalmol 2014; 8: 2319-2323
- 438 Almater A, Alfaleh A, Alshomar K et al. Retinoblastoma: Update on Current Management, Retinoblastoma - Past, Present and Future. In Alkatan H, Hrsg.Retinoblastoma: IntechOpen; 2019
- 439 Cassoux N, Lumbroso L, Levy-Gabriel C. et al. Retinoblastoma: Update on Current Management. Asia-Pac. J Ophthalmol 2017; 6: 290-295
- 440 WHO. RETINOBLASTOMA - 2014 Review of Cancer Medicines on the WHO List of Essential Medicines. In: Union for International Cancer Control; 2014
- 441 Caignard A, Faguer R, Mercier P. et al. Optic nerve and visual pathways primary glioblastoma treated with radiotherapy and temozolomide chemotherapy. Eur J Ophthalmol 2014; 24: 637-640
- 442 Waheed S, Huang B, Zamora C. Glioblastoma involving the optic nerve. Am J Neuroradiol 2018; 41: e
- 443 Alireza M, Amelot A, Chauvet D. Poor prognosis and challenging treatment of optic nerve malignant gliomas: Literature review and case report series. World Neurosurg 2017; 97: e1-e6
- 444 Ashour O, Stalling M, Ramsey J. et al. Intraocular Medulloepithelioma. RadioGraphics 2018; 38: 194-199
- 445 Verdijk R. On the Classification and Grading of Medulloepithelioma of the Eye. Ocul Oncol Pathol 2016; 2: 190-193
- 446 Hei Y, Kang L, Yang X. Orbital alveolar soft part sarcoma: A report of 8 cases and review of the literature. Oncol Lett 2018; 15: 304-314
- 447 Inci E, Korkut N, Erem M. et al. [Alveolar soft tissue sarcoma]. HNO 2004; 52: 145-149
- 448 Rose A, Kabiru J, Rose G. Alveolar soft-part sarcoma of the orbit. Afr J Paediatr Surg 2011; 8: 82-84
- 449 Dirksen U, Jürgens H. S1-Leitlinie: Ewing-Sarkome des Kindes- und Jugendalters. Stand 06/2014, DOI: AWMF-Register Nr. 025/006
- 450 Kaliki S, Rathi S, Palkonda V. Primary orbital Ewing sarcoma family of tumors: a study of 12 cases. Eye 2018; 32: 615-621
- 451 Chaugule S, Putambekar A, Gavade S. et al. Primary Orbital Leiomyosarcoma in an Adult Male. Ophthal Plast Reconstr Surg 2019; 35: e27-e29
- 452 Klippenstein K, Wesley R, Glick A. Orbital Leiomyosarcoma After Retinoblastoma. Ophthalmic Surg Lasers Imaging. Retina 1999; 30: 579-583
- 453 Yeniad B, Tuncer S, Peksayar G. et al. Primary Orbital Leiomyosarcoma. Ophth. Plast Reconstr Surg 2009; 25: 154-155
- 454 Cai Y, McMenamin E, Geoff R. et al. Primary liposarcoma of the orbit: A clinicopathologic study of seven cases. Ann Diagn Pathol 2001; 5: 255-266
- 455 Garrity J, Henderson J, Cameron J. Henderson's Tumors of Primitive Mesoderm, Striated muscle, Adipose Tissue and Smooth muscle.In Garrity JA HJ, Cameron JD , Hrsg. Henderson's orbital tumors. 4th edition. AuflPhiladelphia: Lippincott Williams and Wilkins; 2007: 123–141
- 456 Alkatan H, Chaudhry I, Al-Qahtani A. Epithelioid sarcoma of the orbit. Ann Saudi Med 2011; 31: 187-189
- 457 Inazawa N, Hatakeyama N, Hori T. et al. Primary orbital neuroblastoma in a 1-month-old boy. Pediatr Int 2014; 56: 122-125
- 458 Vallinayagam M, Rao V, Pandian D. et al. Primary orbital neuroblastoma with intraocular extension. Indian J Ophthalmol 2015; 63: 684-686
- 459 Zhang N, Lin L. Presumed primary orbital neuroblastoma in a 20-month-old female. Ophthal Plast Reconstr Surg 2010; 26: 383-385
- 460 Rose A, Luthert P, Jayasena C. et al. Primary Orbital Melanoma: Presentation, Treatment, and Long-term Outcomes for 13 Patients. Front Oncol 2017; 7: 316
- 461 Kiratli H, Balci K, Güler G. Primary orbital endodermal sinus tumor (yolk sac tumor). J AAPOS 2008; 12: 623-625
- 462 Mogaddam A, Memar B, Aledavood A. et al. Isolated Orbital Endodermal Sinus Tumor. Ophthal Plast Reconstr Surg 2007; 23: 477-479
- 463 Eckardt A, Lemound J, Rana M. et al. Orbital lymphoma: diagnostic approach and treatment outcome. World J Surg Oncol. 2013 DOI: 73
- 464 Olsen T, Heegaard S. Orbitla Lymphoma. Surv Ophthalmol 2019; 64: 45-66
- 465 Sharma T, Kamath M. Diagnosis and Management of Orbital Lymphoma. Ophthalmic Pearls. 2015 DOI: 37-39
- 466 Bidar M, Wilson M, Laquis S. et al. Clinical and imaging characteristics of orbital leukemic tumors. Ophthalmic Plast Reconstr Surg 2007; 23: 87-93
- 467 Hatton M, Rubin P. Chronic Lymphocytic Leukemia of the Orbit. Arch Ophthalmol 2002; 120: 990-991
- 468 Maka E, Lukáts O, Tóth J. et al. Orbital tumour as initial manifestation of acute myeloid leukemia: granulocytic sarcoma: case report. Pathol Oncol Res 2008; 14: 209-211
- 469 Taylor C, Taylor R, Kinsey S. LEUKEMIC INFILTRATION OF THE ORBIT: Report of Three Cases and Literature Review. Pediatr Hematol Oncol 2005; 22: 415-422
- 470 Pausch N, Sterker I, Bauer U. Maligner intraorbitaler Tumor: Primum oder Metastase?. HNO 2016; 64: 262-264
- 471 Schenke T, Lehmann G, Kunze C. et al. Eine abszessverdächtige Raumforderung in der extraokulären Augenmuskulatur. HNO. 2020
- 472 Suarez C, Ferlito A, Lund V. Management of the orbit in malignant sinunasal tumors. Head Neck 2008; 30: 242-250
- 473 Evert K, Kühnel T, Weiß K. et al. Diagnostik und Management von Gefäßanomalien. Der. Pathologe 2019; 40: 422-430
- 474 Müller-Wille R, Wildgruber M, Sadick M. et al. Vascular Anomalies (Part II): Interventional Therapy of Peripheral Vascular Malformations. Fortschr Röntgenstr 2018; 190: 927-937
- 475 Sadick M, Müller-Wille R, Wildgruber M. et al. Vascular Anomalies (Part I): Classification and Diagnostics of Vascular Anomalies. Fortschr Röntgenstr 2018; 190: 825-835
- 476 Wassef M, Blei F, Adams D. et al. Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics 2015; 136: e203-e214
- 477 Fay A, Nguyen J, Jakobiec F. et al. Propranolol for Isolated Orbital Infantile Hemangioma. ARCH OPHTHALMOL 2010; 128: 256-258
- 478 Grantzow R, Schmittenbecher P, Höger P. et al. S2k-Leitlinie 006/100: Infantile Hämangiome im Säuglings- und Kleinkindesalter. AWMF online. 2015 DOI: 1-13
- 479 Goldsmith J, van de Rijn M, Syed N. Orbital Hemangiopericytoma and Solitary Fibrous Tumor: A Morphologic Continuum. Internat J Surg Pathol 2001; 9: 295-302
- 480 Pacheco L, Fernandes B, Miyamoto C. et al. Rapid growth of an orbital hemangiopericytoma with atypical histopathological findings. Clin Ophthalmol 2014; 8: 31-33
- 481 Westerfeld C, Allen C, Mihm M. et al. Non-Involuting Congenital Hemangioma Presenting as an Orbital Mass in a Newborn. Investigative Ophthalmology & Visual Science 48: 3582
- 482 Gbolahan O, Fasina O, Adisa A. et al. Spindle cell hemangioma: Unusual presentation of an uncommon tumor. J Oral Maxillofac Pathol 2015; 19: 406
- 483 Budimir I, Demirović A, Iveković R. et al. Epithelioid hemangioma of the orbit: case report. Acta Clin Croat 2015; 54: 92-95
- 484 McEachren T, Brownstein S, Jordan D. et al. Epithelioid hemangioma of the orbit. Ophtalmology 2000; 107: 806-810
- 485 Stiefel H, Ng J, Wilson D. et al. Orbital Cellular Epithelioid Hemangioma. Ocul Oncol Pathol 2019; 5: 424-431
- 486 Kamano H, Noguchi T, Yoshiura T. et al. Intraorbital lobular capillary hemangioma (pyogenic granuloma). Radiat Med 2008; 26: 609-612
- 487 Häußler S, Uecker F, Knopke S. et al. Tufted angiomas of the head and neck. HNO 2018; 66: S1-S6
- 488 Benesch M, Lackner H, Sorantin E. et al. Medikamentöse Therapie vaskulärer Anomalien. Paediatr Paedolog 2020; 55: 21-27
- 489 Cho W, Kim S, Park S. et al. Intracranial kaposiform hemangioendothelioma: proposal of a new malignant variant. J Pediatr Ophthalmol Strabismus 2009; 3: 147-150
- 490 Li B, Li Y, Tian X. et al. Unusual multifocal intraosseous papillary intralymphatic angioendothelioma (Dabska tumor) of facial bones: a case report and review of literature. Diagn Pathol 2013; 8: 160
- 491 Collaco L, Goncalves M, Gomes L. Orbital Kaposi’s Sarcoma in Acquired Immunodeficiency Syndrome. Eur J Ophthalmol 2000; 10: 88-90
- 492 Siddens J, Fishman J, Jackson I. et al. Primary orbital angiosarcoma: a case report. Ophthalmic Plast Reconstr Surg 1999; 15: 454-459
- 493 Lyon D, Tang T, Kidder T. Epithelioid Hemangioendothelioma of the Orbital Bones. Ophtalmology 1992; 99: 1773-1778
- 494 ISSVA. ISSVA Classification of Vascular Anomalies ©2018. In: International Society for the Study of Vascular Anomalies
- 495 Wildgruber M, Sadick M, Muller-Wille R. et al. Vascular tumors in infants and adolescents. Insights Imaging 2019; 10: 30
- 496 Gilbert P, Dubois J, Giroux F. New Treatment Approaches to Arteriovenous Malformations. Semin Intervent Radiol 2017; 34: 258-271
- 497 Schobinger RA, Fontaine R. Periphere Angiodysplasien.





























