2
Outlines of the Reactions Involving SO2 Insertion
2.1
Transition-Metal-Catalyzed SO2 Insertion
Although several methodologies have been developed for SO2 insertions, the transition-metal-catalyzed SO2 insertion is still in demand due to its remarkable catalytic activity and better selectivity.[23]
Sulfonamides are found in many pharmaceuticals and biologically active compounds and hence the development of new methodologies is deemed worthy.[24] A three-component reaction of an arylboronic acid, nitroarene (1), and potassium metabisulfite under copper catalysis was established by Wu et al. yielding a variety of sulfonamides. Various functional groups including hydroxy-, cyano-, amino- and carbonyl were well tolerated in this strategy (Scheme [1]).[25]
 Scheme 1 Synthesis of phenyl N-aryl sulfonamides using nitrobenzene and boronic acids
Scheme 1 Synthesis of phenyl N-aryl sulfonamides using nitrobenzene and boronic acids
According to the mechanism (Scheme [2]), the copper-catalyzed addition of K2S2O5 with an aryl boronic acid generates an arylsulfinate intermediate (d). Further, a copper-assisted nucleophilic interaction of intermediate (e) with nitroarene (1) gives rise to intermediate (1e) followed by protonation to afford intermediate (1f). The intermediate (1f) undergoes reduction with isopropanol, producing a sulfonyl hydroxylamine (1g), which, on further reduction, affords the final product 2.
 Scheme 2 Proposed mechanism for the formation of phenyl N-aryl sulfonamide derivatives
Scheme 2 Proposed mechanism for the formation of phenyl N-aryl sulfonamide derivatives
A similar approach was reported by Wu et al. for the synthesis of sulfonamides 4, which proceeds via a Pd-catalyzed coupling of aryl halides 3, hydrazines, and potassium metabisulfite. Both aryl iodides, as well as aryl bromides, reacted smoothly under identical reaction conditions. However, aryl chlorides and alkyl halides were demonstrated not to be substrates in this conversion (Scheme [3]).[26]
 Scheme 3 Synthesis of sulfonamides using aryl halides
Scheme 3 Synthesis of sulfonamides using aryl halides
In 2019, the Ramon group demonstrated a copper-catalyzed synthesis of sulfonamides 6 from triaryl bismuthines 5, sodium metabisulfite, and nitro compounds as the amino source in a deep eutectic solvent. It was found that substrates containing neutral, electron-withdrawing, and electron-donating substituents all gave products in moderate to good yields. The reaction was successful for the aliphatic nitrocyclohexane, although a lower yield of the desired product was obtained (Scheme [4]).[27]
 Scheme 4 Synthesis of phenyl N-aryl sulfonamide derivatives using nitrobenzene
Scheme 4 Synthesis of phenyl N-aryl sulfonamide derivatives using nitrobenzene
Manabe et al. in 2017, demonstrated an elegant method for the synthesis of sulfonamides 8 and sulfinamides 8′ from heteroarenes 7 bearing an amino group and K2S2O5 as the SO2 surrogate. In this protocol, the selectivity is governed by the nature of the ligand and the equivalents of the base used. The protocol covers a wide range of cyclic amines with good functional group tolerance. From the mechanistic investigations, the group confirmed the generation of sulfonamides, which are converted into sulfonamides in the presence of ≤1.0 equiv of the base. In the process, sulfinamides are produced through an unprecedented insertion of sulfur monoxide, and products were obtained via oxidation using an iodide/DMSO combination. The presence of iodide and DMSO is vital for the successful conversion of sulfinamides into sulfonamides (Scheme [5]).[28]
 Scheme 5 Synthesis of cyclic sulfonamides
Scheme 5 Synthesis of cyclic sulfonamides
Recently, Jiang and co-workers reported a similar multi-component approach for the reductive coupling of sodium metabisulfite (Na2S2O5), 4-iodotoluene (9), and n-butyl bromide in the presence of a Pd-catalyst and potassium hydrogen phosphate. Important features of this protocol are the broad substrate scope, tolerance of various functional groups, simple and cheap coupling partner, and high yields of the products (Scheme [6]).[29]
 Scheme 6 Synthesis of sulfonyl derivatives using alkyl bromides as the coupling partner
Scheme 6 Synthesis of sulfonyl derivatives using alkyl bromides as the coupling partner
From the control experiments performed, a suitable mechanism was proposed. Initially, the n-butyl radical is generated via a single-electron transfer between alkyl halide and tin, which reacts with sodium metabisulfite to give sulfonyl intermediate (a). The intermediate (a) undergoes reduction with tin, giving sulfonyl anion intermediate (b). Intermediate (b) then reacts with intermediate 9a (generated via the oxidative addition of Pd(0) and aryl halide) to give intermediate 9c. Reductive elimination of intermediate 9c provides the sulfonylated product 10 (Scheme [7]).
 Scheme 7 Proposed mechanism for the formation of aryl alkyl sulfones
Scheme 7 Proposed mechanism for the formation of aryl alkyl sulfones
Since methyl sulfones are privileged scaffolds in many pharmaceuticals, synthesis of such molecules has attracted considerable attention.[21] For example, Vismodegib® (Figure [1]) is a basal-cell carcinoma treatment that was first explored by Roche. Xiidra® (Figure [2]) has been applied for dry eye disease as an ophthalmic solution.[30]
 Figure 2 Sulfone-containing drug Xiidra®
Figure 2 Sulfone-containing drug Xiidra®
An elegant synthesis of β-methylsulfonylated N-heterocycles (12 or 12′) via FeCl3-catalyzed C(sp3)–H dehydrogenation and C(sp2)–H methylsulfonylation of unactivated cyclic amines using sodium metabisulfite and dicumyl peroxide (DCP) has been demonstrated by the Fan group. However, in this method, DCP serves as an oxidant as well as a methyl radical source to generate a methyl sulfonyl radical. This protocol provided several β-methylsulfonylated tetrahydropyridines, tetrahydroazepines, and pyrroles in one-pot (Scheme [8]).[31]
 Scheme 8 Synthesis of β-methylsulfonylated N-heterocycles
Scheme 8 Synthesis of β-methylsulfonylated N-heterocycles
In the proposed mechanism, the methyl radical initially generated from DCP is captured by sulfur dioxide to give a methylsulfonyl radical (a) along with the formation of acetone. Meanwhile, compound 11 is oxidized by Fe(III) to deliver a radical cation intermediate 11a, which undergoes dehydrogenation to produce an iminium intermediate 11b and PhC(Me)2OH. Subsequently, enamine intermediate 11c is generated via β-hydrogen abstraction by DABCO or PhC(Me)2O–. Next, the methylsulfonyl radical intermediate A undergoes addition with the enamine intermediate 11c to provide intermediate 11d, which, upon Fe(III)-promoted oxidation, gives cationic species 11e. The final product 12 is obtained upon loss of a proton (Scheme [9]).
 Scheme 9 Proposed mechanism for the formation of aryl alkyl sulfones
Scheme 9 Proposed mechanism for the formation of aryl alkyl sulfones
Similarly, Jiang et al. demonstrated an efficient method for the synthesis of methyl sulfones 14 involving a three-component cross-coupling protocol of boronic acid 13, sodium metabisulfite, and dimethyl carbonate. Important features of the reaction include a wide range of substrate scope, and good functional group tolerance Among the various ligands tested, it was found that electron-rich and sterically hindered phosphine ligands are more suitable for the desired conversion (Scheme [10]).[32]
 Scheme 10 Synthesis of sulfonyl derivatives using dimethyl carbonate as the methylating agent
Scheme 10 Synthesis of sulfonyl derivatives using dimethyl carbonate as the methylating agent
Synthesis of o-substituted diaryl sulfones 16 via a multicomponent reaction of arylboronic acid 15, potassium metabisulfite, and diaryliodonium salt was demonstrated by Tu et al. in 2019 using an acenaphthoimidazolylidene gold complex as the catalyst (Scheme [11]).[33] The sterically hindered aryl groups in diaryliodonium salts are preferentially transformed over less bulky ones during the process, which might be due to the better stability of the bulky Ar+ formed from diaryliodonium salt. A wide variety of diaryl sulfones could be obtained using various arylboronic acids and aryldiazonium salts.
 Scheme 11 Synthesis of diaryl sulfones using boronic acid and diaryliodonium salts
Scheme 11 Synthesis of diaryl sulfones using boronic acid and diaryliodonium salts
According to the proposed mechanism, initial transmetalation of NHC-Au(I) with arylboronic acid 15 provides NHC-Au-Ar species 15a. This is then followed by the insertion of SO2 to provide a sulfonyl Au(I) complex 15b. The NHC-AuSO2-Ar (15b), furnishes an aryl sulfonyl radical intermediate 15c in the presence of base. The combination of intermediate 15c with the more stable bulky Ar+ species (a), generated from diaryliodonium salt, affords the sterically hindered diaryl sulfone 16 (Scheme [12]).
 Scheme 12 Proposed mechanism for the formation of diaryl sulfones
Scheme 12 Proposed mechanism for the formation of diaryl sulfones
By utilizing the same SO2 insertion strategy, the Jiang group reported a method for the synthesis of diarylannulated sulfones 18 and 18′ using Na2S2O5 as SO2 surrogate. The diarylannulated sulfones were synthesized via SO2/I exchange of iodonium (III) salts 17. By this protocol, a new type of OLED material was synthesized on gram scale with good functional group tolerance, permitting a broad range of substrate scope (Scheme [13]).[34]
 Scheme 13 Synthesis of diaryl annulated sulfones
Scheme 13 Synthesis of diaryl annulated sulfones
Often, compounds having a furan2(5H)-one backbone have high biological activity.[35a] For example, Rofecoxib® (Figure [3]) is an anti-inflammatory drug launched by Merck and approved by the US FDA.[35b] Several 4-aryl-3methyl-furan-2(5H)-ones are effective in controlling fungal diseases in plants of agronomic importance.[36] In this context, Wu et al. in 2019 demonstrated a method in which 4-sulfonylated furan-2(5H)-ones 20 are formed by a three-component reaction of 2,3-allenoic acids 19, sulfur dioxide, and aryldiazonium tetrafluoroborates in the presence of a copper catalyst. The method utilizes both DABSO and Na2S2O5 as the SO2 source. Mild reaction conditions, as well as tolerance of various functional groups, such as nitro groups and esters, as well as broad substrate scope are the important features of the protocol. However, steric effects in the aryl diazonium salt greatly affect the outcome, giving a downward trend in the yield of the product (Scheme [14]).[37]
 Figure 3 Rofecoxib® an anti-inflammatory drug
Figure 3 Rofecoxib® an anti-inflammatory drug
 Scheme 14 Synthesis of 4-sulfonylated furan-2(5H)-ones
Scheme 14 Synthesis of 4-sulfonylated furan-2(5H)-ones
Based on literature precedent, the proposed mechanism involves the generation of an aryl radical via single-electron transfer of an aryl diazonium tetrafluoroborate with Cu(I). This radical then reacts with SO2 obtained from Na2S2O5, affording an aryl sulfonyl radical intermediate (a). The C-central position of 2,3-allenoic acid 19 is then attacked by the aryl sulfonyl radical (a) to give intermediate 19a, which is transformed into intermediate 19b, assisted by the copper(II) catalyst. Subsequently, the intermediate 19b undergoes intramolecular nucleophilic attack by the carboxylate anion in the presence of a base, leading to 4-sulfonylated furan-2(5H)-one (20) (Scheme [15]).
 Scheme 15 Proposed mechanism for the formation of 4-sulfonylated furan-2(5H)-ones
Scheme 15 Proposed mechanism for the formation of 4-sulfonylated furan-2(5H)-ones
Alkyl nitriles are present in various natural products and pharmaceuticals.[38] Moreover, cyanoalkyl groups can be readily converted into other useful functional groups such as esters, amides, carboxyls, and tetrazoles.[39] Similarly, oxindoles are privileged scaffolds in many drugs and biologically active compounds.[40] Liu’s group demonstrated an iron-catalyzed protocol for the synthesis of cyanoalkyl sulfonylated oxindoles 22 from activated olefins 21 and cyclic keto oximes via C–C single-bond insertion of sulfur dioxide. The method does not require any additional base or oxidant, which is one of the main advantages of the protocol (Scheme [16]).[41]
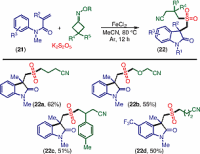 Scheme 16 Synthesis of 3-cyanoalkyl sulfonylated oxindoles
Scheme 16 Synthesis of 3-cyanoalkyl sulfonylated oxindoles
Based on literature precedent and on experimental results, a mechanism for the iron-catalyzed radical cyanoalkylsulfonylation/arylation of active olefins was proposed (Scheme [17]). Initially, the oxime ester (a) undergoes SET reduction by Fe(II) to give an iminyl radical intermediate (b), which forms intermediate (c) via cleavage of the C–C bond. Subsequently, the intermediate (c) is captured by the SO2 generated from K2S2O5, to provide another intermediate (d). Next, the radical intermediate (d) attacks the C–C bond of acrylamide (21) to provide intermediate 21d, which undergoes intramolecular cyclization to give intermediate 21e. Finally, SET oxidation of intermediate 21e by Fe(III) followed by 4-(trifluoromethyl)benzoate ion assisted deprotonation gives the final product 22 and regenerates Fe(II) for the next catalytic cycle.
 Scheme 17 Proposed mechanism for the formation of 3-cyanoalkylsulfonylated oxindoles
Scheme 17 Proposed mechanism for the formation of 3-cyanoalkylsulfonylated oxindoles
In 2021 Yu et al. demonstrated an iron-catalyzed SO2 insertion between 2,3-allenoic acids 23 and cyclobutanone oxime esters using K2S2O5 as the SO2 surrogate (Scheme [18]).[42] During the reaction, ring-opening of the cyclobutanone oxime ester produces a cyanoalkyl radical, which is followed by a radical tandem cyclization providing cyanoalkylsulfonylated butenolides 24.
 Scheme 18 Cyanoalkylsulfonylation of 2,3-allenoic acids
Scheme 18 Cyanoalkylsulfonylation of 2,3-allenoic acids
The suggested mechanism involves the reduction of the cycloketone oxime ester by Fe(II) via SET, leading to the formation of an iminyl radical (a) through N–O bond cleavage. Subsequently, the C–C bond cleavage of intermediate (a) gives an alkyl radical species (b), which, in combination with sulfur dioxide from K2S2O5, provides a sulfonyl radical intermediate (c). This is then followed by the addition to allenoic acid 23 to form intermediate 23c. The intermediate 23c undergoes oxidation in the presence of the Fe(III) catalyst to provide allylic cation 23d. Finally, the cyclized product 24 is obtained via intramolecular nucleophilic attack (Scheme [19]).
 Scheme 19 Proposed mechanism for the cyanoalkylsulfonylation of 2,3-allenoic acids
Scheme 19 Proposed mechanism for the cyanoalkylsulfonylation of 2,3-allenoic acids
2.2
Transition-Metal-Free SO2 Insertion
Although transition-metal-catalyzed SO2 insertion has gained considerable attention, the development of efficient and practical protocols for the direct introduction of sulfonyl group in the absence of a transition-metal catalyst is an attractive approach. The introduction of a sulfonyl group under transition-metal-free conditions is a challenging task.[43]
A metal-free multi-component strategy was disclosed by Wu et al. for the synthesis of nitrile-containing sulfones 26 using aryldiazonium tetrafluoroborates 25, and 3-azido-2-methylbut-3-en-2-ol with sodium metabisulfite as the sulfur dioxide surrogate (Scheme [20]).[44]
 Scheme 20 Synthesis of nitrile-containing sulfones
Scheme 20 Synthesis of nitrile-containing sulfones
A plausible mechanism for this sulfonylation process is described in Scheme [21]. Initially, aryl sulfonyl radical 25a is generated in situ by the reaction of aryldiazonium tetrafluoroborate 25 and sodium metabisulfite. Further, the addition of aryl sulfonyl radical 25a to 3-azido-2-methylbut-3-en-2-ol gives the radical intermediate 25b, which subsequently releases N2, generating a nitrogen-centered radical 25c. The radical intermediate 25c provides arylsulfonylacetonitrile 26 via C–C bond cleavage, along with a ketyl radical (a), which, upon loss of a proton, forms acetone as the sole by-product.
 Scheme 21 Proposed mechanism for the formation of nitrile-containing sulfones
Scheme 21 Proposed mechanism for the formation of nitrile-containing sulfones
Vinyl sulfones are found in many natural products and pharmaceuticals.[45] The reactivity of α,β-unsaturated sulfones leads to various organic transformations via nucleophilic addition, radical addition, and cycloaddition. A metal-free, three-component reaction protocol involving propargyl alcohol 27, sodium metabisulfite, and aryldiazonium tetrafluoroborates was demonstrated by Wu et al. in 2020 (Scheme [22]).[46] The reaction proceeds efficiently at room temperature in the absence of catalyst, providing E-vinyl sulfones 28 in moderate to good yields. The approach involves a vinyl radical-induced 1,5-hydrogen atom transfer and functional group migration, resulting in sequential cleavage of inert C–H and C–C bonds, respectively. Aryldiazonium tetrafluoroborates bearing electron-donating or electron-withdrawing groups on the aromatic ring worked smoothly in this transformation, providing the desired products 28 in moderate to good yields. Besides this, a wide variety of propargyl alcohols was also successful (Scheme [22]).
 Scheme 22 Synthesis of vinyl sulfones
Scheme 22 Synthesis of vinyl sulfones
In the proposed mechanism, an aryl sulfonyl radical intermediate (a) is generated from the aryl diazonium salt and Na2S2O5. Radical addition of intermediate (a) to alkyne 27 provides intermediate 27a, which, upon 1,5-[H] shift, gives intermediate 27b. The intermediate 27b undergoes radical cyclization followed by SET and deprotonation to give product 28 (Scheme [23]).
 Scheme 23 A tentative mechanism for the formation of vinyl sulfones
Scheme 23 A tentative mechanism for the formation of vinyl sulfones
Recently, an elegant method for the synthesis of aryl sulfones was reported by Wu et al. The method offers a range of (arylsulfonyl)methylbenzenes 30 via a multicomponent reaction of electron-rich arenes 29, potassium metabisulfite, aromatic aldehydes, and aryldiazonium tetrafluoroborates in the presence of formic acid (Scheme [24]).[47] The reaction proceeds very well under mild reaction conditions with broad substrate scope tolerating various functional groups.
 Scheme 24 Synthesis of aryl sulfones using aromatic aldehydes and aryl diazonium salts
Scheme 24 Synthesis of aryl sulfones using aromatic aldehydes and aryl diazonium salts
According to the proposed mechanism, condensation of 1,3,5-trimethoxybenzene (29) with an aldehyde in the presence of formic acid generates cationic intermediate 29a. Aryldiazonium tetrafluoroborate reacts with potassium metabisulfite, leading to an arylsulfonyl radical intermediate (b) that attacks intermediate 29a to provide a radical cation 29c. Subsequently, deprotonation via SET affords product 30 (Scheme [25]).
 Scheme 25 A plausible mechanism for the formation of aryl sulfones
Scheme 25 A plausible mechanism for the formation of aryl sulfones
Five-membered sulfur heterocycles have a major presence in medicinal chemistry and materials sciences.[48] In this regard, Larionov et al. in 2018 reported an efficient method for the synthesis of 3-sulfolenes 32 or 32′ from 1,3-dienes 31 or allylic alcohols 31′ with sodium metabisulfite as the SO2 surrogate. Most of the sulfolenes were obtained in good to excellent yields when carried out in HFIP or with KHSO4 in methanol. Broad substrate scope, good product yield, gram-scale synthesis, and metal-free conditions are some noteworthy features of this protocol (Scheme [26]).[49]
 Scheme 26 Synthesis of 3-sulfolenes using 1,3-dienes
Scheme 26 Synthesis of 3-sulfolenes using 1,3-dienes
Sulfonyl fluorides have gained importance and attracted attention due to their unique reactivity and stability. In addition, sulfonyl fluorides are also used in place of sulfonyl chloride for the synthesis of sulfonylated compounds.[50] Inspired by this, Lu and co-workers established a method for the formation of aryl sulfonyl fluorides 34 from arene diazonium salts 33 using sodium metabisulfite as the SO2 source (Scheme [27]).[51] Aryl diazonium salts possessing electron-donating, as well as electron-withdrawing substituents, performed well in this transformation. Furthermore, several heteroaromatic diazo compounds reacted smoothly under identical reaction conditions to give the corresponding sulfonyl fluorides. Using this protocol, a copper-free Sandmeyer fluorosulfonylation was established.
 Scheme 27 Synthesis of aryl sulfonyl fluorides
Scheme 27 Synthesis of aryl sulfonyl fluorides
According to the proposed mechanism, the aryl diazonium salt undergoes SET to give an aryl radical 33a that then captures SO2 from the sodium metabisulfite to give the aryl sulfonyl radical intermediate 33b. The radical intermediate 33b then reacts with the fluoride radical obtained from Selectfluor®, affording the desired product 34. In this protocol, sodium metabisulfite serves the dual role of reductant and SO2 source, enabling this copper-free Sandmeyer-type fluorosulfonylation (Scheme [28]).
 Scheme 28 Proposed mechanism for the formation of aryl sulfonyl fluorides
Scheme 28 Proposed mechanism for the formation of aryl sulfonyl fluorides
Radical difunctionalization of alkenes and alkynes is an interesting approach to introduce two functional groups simultaneously.[52] Singh et al. in 2020 reported an efficient method for the synthesis of β-ketosulfones 36 and 36′ via a multicomponent reaction of alkenes or alkynes, aryldiazonium salts 35, and SO2 derived from K2S2O5 under transition-metal-free conditions. The strategy is equally successful for phenyl acetylenes and styrenes bearing electron-withdrawing as well as electron-donating groups (Scheme [29]).[53]
 Scheme 29 Synthesis of β-ketosulfones
Scheme 29 Synthesis of β-ketosulfones
Similarly, Wu et al. reported a four-component reaction of aryldiazonium salts 37, sulfur dioxide, alkenes, and hydroxylamine. The methodology utilized both DABCO∙(SO2)2 and K2S2O5 as the SO2 surrogate, which underwent a smooth reaction both with aryl diazonium salts and styrenes (Scheme [30]).[54]
 Scheme 30
Vicinal difunctionalization of alkenes via SO2 insertion
Scheme 30
Vicinal difunctionalization of alkenes via SO2 insertion
Sulfonamide synthesis via a metal-free approach is challenging for synthetic chemists. In this regard, Wu et al. in 2021 developed a multicomponent reaction involving nitroarenes 39, arylboronic acids, and potassium metabisulfite, leading to the formation of sulfonamides 40. A noteworthy feature of this protocol is that it is transition-metal-free, and exhibits broad functional group tolerance. A range of sulfonamides bearing different reactive functional groups was obtained in good to excellent yields (Scheme [31]).[55]
 Scheme 31 Metal-free synthesis of sulfonamides
Scheme 31 Metal-free synthesis of sulfonamides
The mechanism shown in Scheme [32] involves decomposition of K2S2O5, resulting in formation of SO2, which undergoes nucleophilic addition with the arylboronic acid to form benzenesulfinate (a). Subsequently, nucleophilic addition of (a) to nitroarene 39 generates intermediate 39a, which couples with another molecule of SO2 to provide intermediate 39b. In the presence of K2CO3, intermediate 39b eliminates SO4
2– to give intermediate 39c. Subsequently, addition of SO2 followed by elimination of SO4
2– provides the desired sulfonamide 40 via the intermediacy of 39d.
 Scheme 32 Proposed mechanism for the formation of sulfonamides
Scheme 32 Proposed mechanism for the formation of sulfonamides
Quinolines are an important class of heterocycles with diverse biological and pharmacological properties.[56] In this context, quinoline N-oxides are valuable starting materials for different transformations to provide functionalized quinolines. Recently, Xia and co-workers developed a metal-free, three-component reaction of quinoline N-oxides 41, sodium metabisulfite, and aryldiazonium tetrafluoroborates via a radical process to give 2-sulfonyl quinolones/ isoquinolines 42 (Scheme [33]).[57] In this approach, aryldiazonium tetrafluoroborates bearing p-substituents were more efficient than those bearing m- and o-substituents. This might be due to steric effects. On the other hand, aryldiazonium tetrafluoroborates bearing electron-donating groups reacted better than those bearing electron-withdrawing groups. Besides this, a variety of quinoline N-oxides and isoquinoline N-oxides worked well in this methodology.
 Scheme 33 Metal-free synthesis of 2-sulfonyl quinolones/isoquinolines
Scheme 33 Metal-free synthesis of 2-sulfonyl quinolones/isoquinolines
In the proposed mechanism (Scheme [34]), the aryldiazonium tetrafluoroborate undergoes thermal decomposition to give an aryl radical (a) that combines with SO2 obtained from sodium metabisulfite to provide an aryl sulfonyl radical (b). Then addition of (b) to quinoline N-oxide 41 via a Minisci-like radical transformation generates an O-radical intermediate 41b that captures another aryl sulfonyl radical to give intermediate 41c. Finally, elimination of aryl sulfonic acid from intermediate 41c affords the corresponding 2-sulfonyl quinolines 42.
 Scheme 34 Proposed mechanism for the formation of 2-sulfonyl quinolones
Scheme 34 Proposed mechanism for the formation of 2-sulfonyl quinolones
In 2020, Ramon et al. developed a catalyst-free methodology for the multicomponent synthesis of sulfones, disulfides, and sulfides using non-toxic triarylbismuthines (Ar3Bi) (43) and sodium metabisulfite in a deep eutectic solvent (DES) (Scheme [35]).[58] The use of DES helped to solubilize all reagents, thereby, enhancing their reactivity. A variety of electrophiles and nucleophiles in the synthesis of sulfones and sulfide worked well.
 Scheme 35 Synthesis of sulfones and sulfides in deep eutectic solvents
Scheme 35 Synthesis of sulfones and sulfides in deep eutectic solvents
In 2018, Jiang et al. demonstrated a metal-free synthesis of sulfonamides 46 via a three-component reaction involving sodium metabisulfite, sodium azide, and aryl diazonium salts 45 (Scheme [36]).[59]
 Scheme 36 Synthesis of primary sulfonamides
Scheme 36 Synthesis of primary sulfonamides
The mechanism for the formation of the product 46 is depicted in Scheme [37]. The process involves the generation of an aryl radical intermediate (a) via SET between the aryl diazonium salt and triphenylphosphine. Then the SO2 combines with the aryl radical intermediate (a) to give an aryl sulfonyl intermediate 45a, which reacts with the phosphine imine radical (b) and sodium azide to provide intermediate 45c. Subsequent hydrolysis of intermediate 45c affords the product 46 along with the by-product triphenylphosphine oxide.
 Scheme 37 Proposed mechanism for the formation of primary sulfonamides
Scheme 37 Proposed mechanism for the formation of primary sulfonamides
Previously, the Pan group in 2013 demonstrated a catalyst-free method for the synthesis of sulfonylhydrazides 48 from phenylhydrazines 47 as the aryl source and potassium metabisulfite as the SO2 source. Metal-free, additive-free conditions, readily available starting materials, and low reaction temperatures are the merits of this protocol, in which phenylhydrazines bearing both electron-withdrawing and electron-donating groups were well tolerated (Scheme [38]).[60]
 Scheme 38 Synthesis of primary sulfonylhydrazides
Scheme 38 Synthesis of primary sulfonylhydrazides
In the suggested mechanism, in the presence of O2, phenylhydrazines 47 undergoes two-step deprotonation to give an aryl radical intermediate 47c via the intermediacy of 47a and 47b. Then the intermediate 47c combines with the sulfonyl anionic intermediate (e) (obtained from the reaction of cyclic amine (d) and K2S2O5) to generate an anionic intermediate 47f along with a radical cation 47h via SET. Oxidation of intermediate 47f affords the aryl sulfone radical 47g, and deprotonation of the intermediate 47h provides intermediate 47i. Finally, radical coupling of intermediate 47g and 47i affords the desired product 48 (Scheme [39]).
 Scheme 39 Proposed mechanism for the formation of sufonylhydrazides
Scheme 39 Proposed mechanism for the formation of sufonylhydrazides
In 2018 Jiang et al. developed an efficient method for the synthesis of sulfonamides 50 from readily available nitrobenzenes 49, boronic acids, and Na2S2O5 as the SO2 surrogate. In this protocol sodium metabisulfite (Na2S2O5) serves the role of both activator as well as reductant during sulfonamidation (Scheme [40]).[61]
 Scheme 40 Synthesis of sulfonamides using nitrobenzenes, boronic acids, and Na2S2O5
Scheme 40 Synthesis of sulfonamides using nitrobenzenes, boronic acids, and Na2S2O5
In the proposed reaction, nitrobenzene (49) reacts with sodium metabisulfite to give intermediate 49a, which is converted into nitrosyl intermediate 49b with the release of Na2SO3 and SO3 (Scheme [41]). Simultaneously, sodium metabisulfite acts as an anionic counterpart to activate the C–B bond of boronic acid (a) to afford an SO2 conjugate intermediate (b). The sulfonyl radical intermediate (c) is generated from (b) via 1,5-migration and SET. Subsequently, intermediate 49b and (c) combine to give the nitroso radical intermediate 49d followed by hydrolysis to afford the desired product 50.
 Scheme 41 Proposed mechanism for the formation of N-aryl sulfonamides
Scheme 41 Proposed mechanism for the formation of N-aryl sulfonamides
In 2020, the Shun-Jun Ji group reported an efficient TFA-promoted, transition-metal-free, multicomponent reaction of aryldiazonium salts 51 with sodium metabisulfite (Na2S2O5) and thiols to construct thiosulfonates 52 (Scheme [42]).[62] The reaction proceeds smoothly with a broad tolerance of functional groups present in the aromatic rings of both the aryldiazonium salts as well as in thiols. Moreover, heteroaromatic and aliphatic thiols are also well tolerated to afford the desired thiosulfonates.
 Scheme 42 Synthesis of thiosulfonates
Scheme 42 Synthesis of thiosulfonates
Based on the proposed mechanism (Scheme [43]) TFA reacts with diazonium salt 51 to generate the corresponding aryl radical 51a. This aryl radical 51a then reacts with Na2S2O5 to give an arylsulfonyl radical intermediate 51b that combines with the sulfur anion to give radical anion intermediate 51c. The radical anion 51c undergoes SET to give the thiosulfonate 52.
 Scheme 43 Proposed mechanism for the formation of thiosulfonates
Scheme 43 Proposed mechanism for the formation of thiosulfonates
2.3
Visible-Light-Mediated SO2 Insertion
Recently, visible-light-mediated functionalizations have emerged as significant methodologies in contemporary organic chemistry.[63]
[64] However, most organic compounds do not absorb visible light efficiently, which limits the application of light-mediated organic synthesis. To overcome this, either a transition-metal complex (complexes of Rh, Ir, Ru) or organic dyes such as eosin Y or rose Bengal are used as sensitizers for the required photochemical transformation.[65,66] As mentioned above, various methods used for SO2 insertion require high temperatures and harsh reaction conditions; hence, there is a need for developing methods for SO2 insertion under milder reaction conditions. In this context, SO2 insertion reaction mediated by visible light either in the presence of transition-metal complexes or organic dyes as photoredox catalysts has gained a place in organic synthesis.[67]
[68]
In 2020 Wu et al. reported a photocatalytic synthesis of alkylalkynyl sulfones 54 through the insertion of sulfur dioxide between potassium alkyltrifluoroborates 53 and alkynyl bromides using sodium metabisulfite (Na2S2O5) as the SO2 source (Scheme [44]).[69] This photoinduced reaction proceeded well at room temperature, had broad substrate scope, and gave the products in moderate to good yields.
 Scheme 44 Visible-light-mediated synthesis of alkylalkynyl sulfones
Scheme 44 Visible-light-mediated synthesis of alkylalkynyl sulfones
According to the proposed mechanism (Scheme [45]) an alkyl radical 53a is generated from potassium alkyltrifluoroborate 53 by the influence of the photocatalyst under visible-light irradiation. Subsequently, the sulfur dioxide generated from Na2S2O5 couples with radical 53a to provide alkylsulfonyl radical 53b that then attacks the C–C triple bond of the alkynyl bromide to provide vinyl radical intermediate 53c. Next, SET converts vinyl radical 53c into vinyl anion 53d. Finally, elimination of bromide gives rise to alkylalkynyl sulfone 54.
 Scheme 45 Proposed mechanism for the formation of alkylalkynyl sulfones
Scheme 45 Proposed mechanism for the formation of alkylalkynyl sulfones
Thiosulfonates are useful building blocks in organic synthesis owing to their unusual reactivity and stability.[70] A visible-light-promoted synthesis of thiosulfonates 56 was reported by He et al. using thiols, aryldiazonium salts 55, and sodium metabisulfite under metal-free conditions (Scheme [46]).[71] This mild, three-component reaction utilizes rhodamine 6G as the photocatalyst to provide unsymmetrical thiosulfonates.
 Scheme 46 Visible-light-mediated synthesis of thiosulfonates
Scheme 46 Visible-light-mediated synthesis of thiosulfonates
Following the proposed mechanism (Scheme [47]), rhodamine 6G (PC) is first excited to an excited species PC* under visible-light irradiation. Then, aryl radical 55a is generated from the aryldiazonium salt 55 via SET with the release of N2 and BF4
−. Subsequently, the interaction of aryl radical 55a with Na2S2O5 generates arylsulfonyl radical 55b and Na2SO3. The PC radical cation obtained from the SET process oxidizes the thiol to produce a thiyl radical cation, which is deprotonated by BF4
− to produce a thiyl radical (c). Finally, coupling of arylsulfonyl radical 55b with the thiyl radical (c) gave the product 56; whereas homocoupling provided the disulfide.
 Scheme 47 Proposed mechanism for the formation of thiosulfonates
Scheme 47 Proposed mechanism for the formation of thiosulfonates
Subsequently, Wu and co-workers reported a photoinduced three-component reaction of aryldiazonium tetrafluoroborates 57, sodium metabisulfite, and thiourea, leading to S-aryl thiosulfonates 58 (Scheme [48]).[72] A variety of aryldiazonium tetrafluoroborates worked well in this transformation. This method was unsuccessful for S-alkyl thiosulfonate preparation because of stability issues with the alkyl diazonium tetrafluoroborates. Moreover, the reaction was unsuccessful for 2-substituted aryldiazonium tetrafluoroborates. This might be due to the steric hindrance.
 Scheme 48 Visible-light-mediated synthesis of S-aryl thiosulfonates
Scheme 48 Visible-light-mediated synthesis of S-aryl thiosulfonates
According to the suggested mechanism (Scheme [49]), initial reaction between thiourea and aryldiazonium tetrafluoroborate 57 generates a salt intermediate 57a, which reacts with Na2S2O5 to give sodium thiophenolate (d), and urea with the release of SO2. In the presence of visible light, the photocatalyst is exited and produces aryl radical 57b from another molecule of aryldiazonium tetrafluoroborate 57 via SET. Aryl radical 57b then reacts with SO2, generated from sodium metabisulfite, giving aryl sulfonyl radical intermediate 57c. Subsequently, thiophenolate anion (d) affords an aryl sulfur radical (e), regenerating the ground-state photocatalyst. Finally, the combination of arylsulfonyl radical 57c with the aryl sulfur radical (e) affords S-aryl thiosulfonate 58.
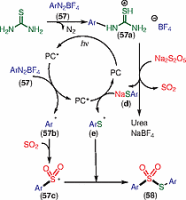 Scheme 49 Proposed mechanism for the synthesis of S-aryl thiosulfonates
Scheme 49 Proposed mechanism for the synthesis of S-aryl thiosulfonates
A Ru(II) photoredox-catalyzed synthesis of α,α-difluoro-β-ketosulfones 60 was reported by Wu et al. in 2020 (Scheme [50]).[73] The reaction involves a three-component coupling of aryldiazonium tetrafluoroborates 59 with sodium metabisulfite and 2,2-difluoroenol silyl ethers under mild conditions. In this conversion, the difluoromethyl group and sulfone moiety can be introduced in a single step.
 Scheme 50 Visible-light-mediated synthesis of α,α-difluoro-β-ketosulfones
Scheme 50 Visible-light-mediated synthesis of α,α-difluoro-β-ketosulfones
Based on the suggested mechanism as depicted in Scheme [51], initially, aryl radical 59a is generated from the aryldiazonium tetrafluoroborate 59 via SET. Intermediate 59a then reacts with SO2, giving rise to arylsulfonyl radical 59b. Intermediate 59b subsequently attacks the double bond of the difluoroenoxysilane to provide a carbon-centered radical intermediate 59c followed by SET with the oxidized photocatalyst, to yield carbocationic intermediate 59d. Finally, the product 60 is formed through a fluoride-mediated desilylation.
 Scheme 51 Proposed mechanism for the formation of α,α-difluoro-β-ketosulfones
Scheme 51 Proposed mechanism for the formation of α,α-difluoro-β-ketosulfones
In 2017, Manolikakes et al. established a method for the formation of sulfonamides 62 by reacting diaryldiazonium salts 61, Na2S2O5, and hydrazines under visible-light photoredox catalysis. In this reaction, a combination of sodium metabisulfite and acid TFA is used as the SO2 surrogate and perylene (PDI) as the photoredox catalyst. Both aromatic as well as aliphatic hydrazines worked well under identical reaction conditions (Scheme [52]).[74]
 Scheme 52 Synthesis of sulfonamide using PDI as photoredox catalyst
Scheme 52 Synthesis of sulfonamide using PDI as photoredox catalyst
In the proposed mechanism, formation of a stable hydrazine-sulfur dioxide adduct (b) is suggested. Meanwhile PDI* is produced by irradiation of photoredox catalyst PDI. The radical quenching of PDI* with the hydrazine-sulfur dioxide complex (b) forms a radical cation (c), which undergoes deprotonation to give a sulfonyl radical intermediate (d). Simultaneously, aryl radical 61b, generated from the aryl diazonium salt 61 via an electron transfer from PDI radical anion, couples with the sulfonyl radical intermediate (d) to provide the final product 62 (Scheme [53]).
 Scheme 53 Proposed mechanism for the formation of sulfonamides
Scheme 53 Proposed mechanism for the formation of sulfonamides
In 2019, Wu et al. demonstrated an efficient method for the synthesis of 3-(methylsulfonyl)benzo[b]thiophenes 64 by reacting methyl(2-alkynyl phenyl)sulfonates 63 with sodium metabisulfite as the SO2 source in the presence of a Ru complex as the photoredox catalyst and sodium methyl sulfinate as the initiator for the reaction. Low catalyst loading, room temperature reaction and broad substrate scope are important features of the reaction. The protocol was successfully applied using methyl(2-alkynyl phenyl)sulfonates 63, bearing electron-donating and electron-withdrawing groups (Scheme [54]).[75]
 Scheme 54 Synthesis of 3-(methylsulfonyl)benzo[b]thiophenes
Scheme 54 Synthesis of 3-(methylsulfonyl)benzo[b]thiophenes
In the proposed mechanism, the excited state of the photocatalyst oxidizes methylsulfinate to a methylsulfonyl radical (a) as an initiator via SET. The triple bond of methyl(2-alkynyl phenyl)sulfonate (63) is attacked by the methylsulfonyl radical (a), providing the cyclized product 64 with the release of a methyl radical. The released methyl radical is subsequently captured by sulfur dioxide, to give the methylsulfonyl radical (a). After this, the methylsulfonyl radical (a) reacts with methyl(2-alkynyl phenyl)sulfonate (63) to give product 64, with regeneration of the methyl radical. In this process, the methyl radical relay combined with the insertion of sulfur dioxide provides a useful route towards methylsulfonyl containing compounds (Scheme [55]).
 Scheme 55 Proposed mechanism for the formation of 3-(methylsulfonyl)benzo[b]thiophenes
Scheme 55 Proposed mechanism for the formation of 3-(methylsulfonyl)benzo[b]thiophenes
Wu et al. reported a UV-irradiation mediated synthesis of allylic sulfones 66 by reacting aryl/alkyl halides 65, potassium metabisulfite, and allylic bromides. The desired transformation was successful without any metal or photoredox catalyst. A broad reaction scope covering alkyl and aryl halides was demonstrated, and various sensitive functional groups such as amino and ester groups were well tolerated (Scheme [56]).[76]
 Scheme 56 Visible-light-mediated synthesis of aryl/alkyl sulfonamides
Scheme 56 Visible-light-mediated synthesis of aryl/alkyl sulfonamides
Following the recent trends in C(sp3)–H functionalization for sulfonylation, the Wu group demonstrated a visible-light-mediated sulfonylation of 2,4,6-trimethylphenol (67) using sodium metabisulfite (Na2S2O5) as the SO2 surrogate. Although the result is interesting, the substrate scope was limited and only 4-methyl phenols with a methyl or tert-butyl group attached to the ortho-position are suitable for this transformation. The reactions failed to provide the desired products when 4-methylphenols with other groups attached to the ortho-position were used. However, benzylic C(sp3)–H bond functionalization was achieved under mild conditions and visible-light irradiation by using this protocol (Scheme [57]).[77]
 Scheme 57 Visible-light-mediated benzylic C(sp3)–H sulfonylation of 2,4,6-trialkylphenols
Scheme 57 Visible-light-mediated benzylic C(sp3)–H sulfonylation of 2,4,6-trialkylphenols
According to the proposed mechanism, an aryl radical (b) is generated from the aryl diazonium salt (a) by the excited Ir(bpy)3, which combines with the sulfonyl radical to provide an arylsulfonyl radical intermediate (c). Meanwhile, phenol 67 undergoes oxidation via a SET to give intermediate 67a, followed by intermolecular hydrogen atom abstraction to give the benzylic radical intermediate 67b. Finally, combination of intermediate 67b and (c) affords the desired product 68 (Scheme [58]).
 Scheme 58 Proposed mechanism for benzylic C(sp3)–H sulfonylation of 2,4,6-trialkylphenols
Scheme 58 Proposed mechanism for benzylic C(sp3)–H sulfonylation of 2,4,6-trialkylphenols
Alkylnitriles are privileged scaffolds in various natural products and pharmaceuticals and can be readily converted into other useful functional groups, including esters, amides, carboxyls, and tetrazoles. In this context, in 2019, the Wu group demonstrated a method for sulfonylation of alkenes 69 using o-acyl oximes in the presence of Ir(bpy)3 as the photoredox catalyst under visible-light irradiation. A wide range of substrates worked well with good functional group tolerance (Scheme [59]).[78]
 Scheme 59 Visible-light-mediated sulfonylation of o-acyl oximes
Scheme 59 Visible-light-mediated sulfonylation of o-acyl oximes
Mechanistically, it was suggested that, in the presence of a photocatalyst and visible light, N–O bond cleavage of O-acyloxime (a) provides an iminyl radical intermediate (b), which undergoes ring opening via C–C bond cleavage to give a carbon-centered radical (c). The latter carbon radical is captured by SO2, providing a sulfonyl radical (d), which, on reaction with alkene 69, provides another carbon-centered radical 69d. Subsequently a cationic intermediate 69e is generated from 69d via SET, and is attacked by the nucleophilic MeOH in the presence of a base to give the desired product 70 (Scheme [60]).
 Scheme 60 Proposed mechanism for sulfonylation of O-acyl oximes
Scheme 60 Proposed mechanism for sulfonylation of O-acyl oximes
In 2020, Tang and co-workers described a similar protocol in which simultaneous cleavage of two C–C bonds of methylenecyclopropane 71 and cycloketone oxime gave 2-cyanoalkylsulfonylated 3,4-dihydronaphthalenes 72 (Scheme [61]).[79]
 Scheme 61 Visible-light-mediated synthesis of 2-cyanoalkylsulfonylated 3,4-dihydronaphthalenes
Scheme 61 Visible-light-mediated synthesis of 2-cyanoalkylsulfonylated 3,4-dihydronaphthalenes
In the suggested mechanism, cycloketone oxime (a) undergoes reduction by the excited state photocatalyst [Ru(bpy)3]2+* providing an iminyl radical (b), which undergoes C–C bond cleavage to give a cyanoalkyl radical (c), which is captured by the SO2 to give a cyanoalkyl sulfonyl radical (d). Both the cyanoalkyl intermediate (c), and cyanoalkylsulfonyl radical intermediate (d) are trapped by 1,1-diphenylethylene (p) to give adducts (q) and (r), respectively. The cyanoalkylsulfonyl radical (d) then adds to the C–C double bond of the methylene cyclopropane 71, providing intermediate 71d. The intermediate 71d undergoes ring-opening via another C–C bond cleavage to provide carbon-centered radical 71e. After this, intermediate 71f is generated by an intramolecular cyclization of intermediate 71e, which undergoes SET from [Ru(bpy)3]3+. Deprotonation of intermediate 71f in the presence of a base affords product 72 and, finally, [Ru(bpy)3]3+ reverts to the ground state [Ru(bpy)3]2+ (Scheme [62]).
 Scheme 62 Mechanism for synthesis of 2-cyanoalkylsulfonylated 3,4-dihydronaphthalenes
Scheme 62 Mechanism for synthesis of 2-cyanoalkylsulfonylated 3,4-dihydronaphthalenes
Recently, N-functionalized pyridinium salts such as Katritzky’s salt have been found to be effective alkylating agents under photoredox catalytic process for various transformations.[80] In 2019, Wu and co-workers reported the synthesis of β-keto sulfone 74 using Katritzky’s salt as an alkyl radical precursor and potassium metabisulfite as the SO2 surrogate (Scheme [63]).[81] A broad reaction scope, good functional group tolerance including amino, cyano, hydroxy, trifluoromethyl groups and good product yields are the merits of this methodology.
 Scheme 63 Visible-light-mediated synthesis of β-keto sulfones
Scheme 63 Visible-light-mediated synthesis of β-keto sulfones
Following the suggested mechanism, a photoredox excited Ir(III) species generates an alkyl radical (c) through an intermediate (b) from the Katritzky salt (a) via SET. Then sulfur dioxide from potassium metabisulfite combines with the alkyl radical intermediate (c) to give an alkylsulfonyl radical intermediate (d) that is trapped by the silyl enol ether 73, leading to a carbon radical intermediate 73d. In the presence of Ir(IV), this carbon radical intermediate is oxidized to a carbocation species 73e that then undergoes desilylation in the presence of base to afford the corresponding β-keto sulfone 74 (Scheme [64]).
 Scheme 64 Mechanism for synthesis of β-keto sulfones
Scheme 64 Mechanism for synthesis of β-keto sulfones
Use of 4-substituted Hantzsch esters as alkyl radical precursors has been demonstrated and these alkyl units could be readily installed into various substrates.[82] The alkyl radical is generated from the 4-alkyl Hantzsch ester under visible-light irradiation in the presence of a photoredox catalyst through SET. Thus, synthesis of alkynyl sulfones 76 involves the reaction of 4-alkyl Hantzsch esters 75, sodium metabisulfite, and alkynyl bromides under metal-free photoinduced conditions as reported by Wu et al. in 2020 (Scheme [65]).[83] This transformation proceeds smoothly under visible-light irradiation at room temperature, giving rise to the corresponding alkyl alkynyl sulfones 76 in moderate to good yields. Besides this, a broad range of substrate scope with good functional group tolerance are other merits of the methodology.
 Scheme 65 Visible-light-mediated synthesis of alkyl alkynyl sulfones
Scheme 65 Visible-light-mediated synthesis of alkyl alkynyl sulfones
According to the proposed mechanism, alkyl radical 75b is generated from the 4-alkyl Hantzsch ester 75 in the presence of the photocatalyst under irradiation. The alkyl sulfonyl radical 75d is formed via trapping of sulfur dioxide by the alkyl radical intermediate 75b. Subsequently, addition of alkyl sulfonyl radical 75d to the alkynyl bromide produces vinyl radical intermediate 75e. With the assistance of excited photocatalyst, vinyl anion 75f is formed, which affords the corresponding alkylalkynyl sulfone 76 with the release of bromide anion (Scheme [66]).
 Scheme 66 Mechanism for the synthesis of alkyl alkenyl sulfones
Scheme 66 Mechanism for the synthesis of alkyl alkenyl sulfones
An elegant method for the synthesis of 2-sulfonyl-substituted 9H-pyrrolo[1,2-a]indoles 78 was reported by Xie et al. in 2019 through reaction of aryldiazonium tetrafluoroborates, potassium metabisulfite, and N-propargylindoles 77 under visible-light irradiation (Scheme [67]).[84] The proposed mechanism involves the generation of an aryl radical (a) from the aryldiazonium tetrafluoroborate by the assistance of [Ru(bpy)3]2+* via SET. The aryl radical (a) then reacts with the potassium metabisulfite and captures SO2 to afford aryl sulfonyl radical (b) that then attacks the triple bond of N-propargylindole 77 giving rise to vinyl radical intermediate 77b. This is followed by intramolecular cyclization and generation of cyclic radical intermediate 77c. This cyclic radical intermediate undergoes oxidative SET and generates cationic intermediate 77d, which undergoes deprotonation and isomerization to afford the desired cyclic product 78 (Scheme [67]).
 Scheme 67 Mechanism for synthesis of 2-sulfonyl-substituted 9H-pyrrolo[1,2-a]indoles
Scheme 67 Mechanism for synthesis of 2-sulfonyl-substituted 9H-pyrrolo[1,2-a]indoles




