
RSS-Feed abonnieren
DOI: 10.1055/a-2347-9919
HIF Stabilizer Desidustat Protects against Complement-Mediated Diseases

Abstract
Complement cascade is a defence mechanism useful for eliminating pathogenic microorganisms and damaged cells. However, activation of alternative complement system can also cause inflammation and promote kidney and retinal disease progression. Inflammation causes tissue hypoxia, which induces hypoxia-inducible factor (HIF) and HIF helps the body to adapt to inflammation. In this study, we investigated the effect of HIF stabilizer desidustat in complement-mediated diseases. Oral administration of desidustat (15 mg/kg) was effective to reduce the kidney injury in mice that was induced by either lipopolysaccharide (LPS), doxorubicin or bovine serum albumin (BSA)-overload. Complement activation-induced membrane attack complex (MAC) formation and factor B activity were also reduced by desidustat treatment. In addition, desidustat was effective against membranous nephropathy caused by cationic BSA and retinal degeneration induced by sodium iodate in mice. C3-deposition, proteinuria, malondialdehyde, and interleukin-1ß were decreased and superoxide dismutase was increased by desidustat treatment in cBSA-induced membranous nephropathy. Desidustat specifically inhibited alternative complement system, without affecting the lectin-, or classical complement pathway. This effect appears to be mediated by inhibition of factor B. These data demonstrate the potential therapeutic value of HIF stabilization by desidustat in treatment of complement-mediated diseases.
#
Introduction
Complement pathway plays an important role in defence against invading pathogens. It has three distinct components, namely the classical, lectin (mannose-binding lectin), and alternative complement systems. Complement protein C3 is the main component of the complement system, which generates active fragments C3a and C3b by proteolytic cleavage process. Complement activation is a cascading event which results in the formation of C5b-9 (membrane attack complex, MAC). This complex aids in the elimination of dead or pathogenic cells through processes such as opsonization, cell recruitment, or cell lysis. Aberrant activation of complement system can cause serious disorders [1], like kidney diseases, namely IgA glomerulonephritis (IgAN), membranous nephropathy (MN), paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and C3 glomerulopathy (C3G), lupus nephritis, and anti-glomerular basement membrane (GBM) disease, and also retinal diseases like macular degeneration [2].
Hypoxia-inducible factors (HIFs) detect changes in cellular oxygen levels and regulate metabolic changes that drive cellular adaptation to low oxygen availability. HIFs are abundantly expressed in inflammatory cells and regulate immunity [3]. Prolyl hydroxylase domain (PHD) enzymes cause the degradation of HIFs (HIF-1, HIF-2, and HIF-3). Inhibition of PHD enzymes can stabilize HIFs, which can stimulate erythropoietin [4]. HIFs can also decrease hepcidin regulating iron availability in the body [5]. Inflammation is known to cause an increase in hepcidin levels [6]. Desidustat, a novel PHD inhibitor, is clinically used for the treatment of chronic kidney disease-associated anemia [7]. Desidustat had demonstrated an anti-inflammatory effect by suppressing proinflammatory cytokines and the recruitment of inflammatory cells [8].
In this work, we have investigated the effect of desidustat, a PHD inhibitor, on complement systems activation and its role in complement-driven membranous nephropathy and retinal degeneration.
#
Materials and Methods
Animals
Female Balb/c mice, and male Sprague Dawley (SD) rats were procured from the animal research facility of Zydus Research Centre and housed in groups at controlled temperature and humidity conditions, with a 12-h light/dark cycle. The animals had access to a standard chow diet and water ad libitum. Animal protocols were approved by the Institutional Animal Ethics Committee of Zydus Research Centre (Ahmedabad, India), a facility accredited by AAALAC International. The mice or rat were bled by retro-orbital puncture under isoflurane anesthesia. All animal experiments are carried out in accordance to National Research Council’s Guideline for animal use [9].
#
Chemicals
Desidustat, factor B substrate-Ac-SHLGLAR-pNA, and cationic bovine serum albumin were synthesized at Zydus Research Centre (ZRC), Ahmedabad, India. For in vivo studies, desidustat was administered orally as a suspension containing 0.5% Tween 80, 0.5% polyethylene glycol 400, and 0.25% carboxymethyl cellulose in water. All other chemicals were procured from Sigma Aldrich, unless otherwise indicated. For alternative complement assay, desidustat was dissolved in DMSO and prepared in gelatin egtazic acid (EGTA) buffer, while for classic and lectin pathways, desidustat was prepared in gelatin veronal buffer (12–624E, Lonza Bioscience). Anti-membrane attached complex antibody C5b-9 (aE11) antibody (sc-58935, Santa Cruz Biotechnology, Inc.), Mouse IgG Horseradish Peroxidase-conjugated Antibody (HAF007, R&D Systems), fluorescein (FITC)–conjugated Affinipure Goat Anti-Mouse IgG (SA00003–1, Proteintech Group, Inc), Mouse/Rat complement component C3d Antibody (AF2655, R&D Systems), Goat IgG HRP-conjugated Antibody (HAF017, R&D Systems), and C3/C3b/C3c Polyclonal antibody (21337–1-AP, Proteintech Group, Inc) were used in the experiments.
#
Ex vivo inhibition of alternative, classical, and lectin complement pathway
Zymosan A from Saccharomyces Cerevisiae (Z4250, Sigma Aldrich), suspension in Tris-buffered saline (TBS, pH-7.6), was activated by boiling in a sealed container at 100°C for 10 minutes in a water bath. The suspension was centrifuged at 4000 rpm for 30 minutes and the pellet containing the activated zymosan A was re-suspended in TBS buffer and stored at − 20°C. ELISA plate was coated with activated zymosan A (1 mg/mL), C1q (12.5 µg/mL, C1740, Sigma Aldrich) and Mannan from Saccharomyces cerevisiae (1 mg/mL, M7504, Sigma Aldrich) in carbonate buffer (pH 9.5) for evaluating alternative, classical, and lectin complement pathways, respectively, and incubated overnight at 2–8°C. Next day, plates were washed by TBS-T (0.1% tween 20 in TBS) and blocked by adding 1% BSA in TBS for 1 h at room temperature, and again washed with TBS-T. Human serum (25% in gelatin-EGTA buffer or in gelatin-veronal buffer) was incubated with different concentrations of desidustat (0.001 µM to 100 µM in EGTA buffer or gelatin-veronal buffer) for 30 min at room temperature in microfuge tubes. EGTA buffer was used for testing alternative complement pathway and gelatin veronal buffer was used for testing classical or lectin complement pathway. These samples were added to ELISA plate and incubated for 1 h at 37°C. Reaction was terminated by aspirating the supernatant, and plate was washed with TBS-T. Primary antibody, C5b-9 (sc-58935, 2000-fold dilution) was added the plates was incubated for 1 h at room temperature. Plate was washed and incubated with HRP (HAF007, 500-fold dilution) for 1 h at room temperature. After washing with TBS-T, TMB substrate was added and plate was incubated for 15 minutes at room temperature. Reaction was stopped by addition of 1 N sulfuric acid and absorbance was detected at 450 and 570 nm. Corrected optical density (450nm–570nm) was used for calculation. EDTA-treated serum was considered as maximum inhibition and EGTA-treated serum was considered as maximum signal. Inhibition was calculated as: % inhibition=[((Maximum avg - blank)-(Test well avg – blank)) x 100]/ (Maximum avg – blank). The sample without serum was considered blank [10].
#
Inhibition of MAC deposition on RBC cell surface
Human serum (50% serum in gelatin-EGTA buffer) was incubated with desidustat (0.1 µM to 10 µM) for 30 min at room temperature in microfuge tubes. Separately, rat whole blood was collected in a tube containing disodium EDTA tube and stored on ice. This rat whole blood sample was washed three times with sodium iodide (0.16 M in normal saline), removing the white buffy coat containing WBC and platelets. RBC suspension (0.1% v/v) was prepared by aspirating cells in EGTA/EDTA buffer. RBC suspension (50 µL) was added to 100 µL of reaction mixture containing human serum with or without desidustat, and it was incubated for 30 min at 37°C. Reaction was terminated by aspirating the supernatant, and cells were washed two times with PBS. Membrane attack complex deposited on rat RBCs were detected by the addition of an anti-C5b-9 antibody (sc-58935, 100X, 30 min incubation on ice). The cells were washed twice with PBS and incubated with secondary antibody, fluorescein (FITC)–conjugated Affinipure Goat Anti-Mouse IgG (SA00003–1, 1000X) for 30 min on ice. These cells were washed twice with PBS. One tube of vehicle control (EGTA buffer control)-not stained with primary or secondary antibody- served as unstained control. MAC deposition was detected using flow cytometry (Cytoflex SRT, Beckman Coulter) with a 488 nm laser for excitation and fluorescence emission was detected using a 525 nm filter.
#
Factor B inhibition
Factor B inhibition was assessed using human and mouse serum (25%) in 100 mM glycine-HCl buffer, pH 9.5. Human or mouse serum was incubated with desidustat for 15 min, and then this reaction mixture was incubated with 100 µM Ac-SHLGLAR-pNA (factor B substrate) at 37°C. The change in absorbance for 30 min at 1 min interval was measured at 405 nm. The rate of reaction was calculated and expressed against vehicle control [11].
#
Lipopolysaccharide-induced complement activation
Female Balb/c mice were treated with desidustat (15 mg/kg, PO), four hours before and after lipopolysaccharide (LPS, 2 mg/kg, IP) challenge. After twenty-four hours of the LPS challenge, mice were bled and ic3b and c3dg were assessed in plasma by western blot. Briefly, plasma samples (2 μg total protein) were loaded for electrophoresis on a 10% polyacrylamide gel. The samples were wet-transferred to 0.45μm nitrocellulose membrane (Bio-rad). Blots were blocked for 1 h with 5% BSA in TBS-T buffer (20 mM Tris–HC, 0.8% NaCl and 0.05% Tween 20, pH 7.6) and incubated overnight at 4°C with a mouse complement component C3d Affinity purified PAb (AF2655, 500-fold dilution). These blots were washed with TBS-T and incubated with secondary rabbit anti-goat IgG HRP affinity purified PAb (HAF017, 1000-fold dilution) for 1 h. Bands were detected using enhanced chemiluminescence and quantified by gel DOCx.
#
LPS-induced lupus nephritis
Female Balb/c mice were treated with vehicle or desidustat (15 mg/kg, PO), thirty min before the LPS (5 mg/kg, IP) challenge, followed by second treatment of vehicle or desidustat six hours later, twice a day for a day. Normal control animals were given normal saline by IP route. Mice were bled retro-orbitally under isoflurane anesthesia 24 h after LPS treatment and serum was separated and analyzed for creatinine and urea.
#
Doxorubicin-induced nephrotoxicity
Female Balb/c mice were treated with doxorubicin (10 mg/kg, intravenous route). Normal control animals were given normal saline by IV route. After 5 weeks of treatment, mice were randomized based on total urinary protein excretion into vehicle control and desidustat (15 mg/kg, once a day), and the treatment continued for two weeks. At the end of treatment, mice were housed individually in metabolic cages for 24-hour urine protein excretion measurement.
#
BSA overload-induced glomerulonephritis
On day 1 to day 5, female Balb/c received intraperitoneal injections of BSA at doses of 2 mg/kg, 4 mg/kg, 6 mg/kg, 8 mg/kg, and 10 mg/kg, respectively. The mice were administered BSA (10 mg/kg) for next 5 days. Normal control animals were given drinking water by PO route. The animals received either a vehicle or desidustat (15 mg/kg, once a day by oral route) from day 1 to day 10. On the 11th day, the mice were kept in metabolic cages for 24-hour urine collection and total protein excretion was measured.
#
Cationic BSA-induced membranous nephropathy
Cationic BSA (cBSA) was prepared as described in the literature [12]. Female Balb/c mice were immunized by subcutaneous (SC) injection of 0.2 mg of emulsified cBSA in complete Freund’s adjuvant (1:1). On 15th, 17th, 19th and 21st day after immunization, mice were challenged with 50, 100, 200 and 400 µg of cBSA (per mouse) by SC injection, respectively. Normal control animals were given normal saline by SC route. Starting from the 23rd day, these mice were given 400 µg of cBSA per animal by SC injection every other day for the next five weeks. Desidustat (15 mg/kg, once a day) treatment was started from the day of the challenge (15th day) for next six weeks. At the end of the treatment, mice were kept in metabolic cages and 24-hour urine protein excretion was measured. After euthanization, one of the kidneys was collected in 10% formalin for histological assessment and other kidney was dissected and stored in liquid nitrogen.
#
Sodium iodate induced retinal degeneration
Male SD rats were treated with a single dose of sodium iodate (80 mg/kg, IV). These rats were then treated with desidustat (15 mg/kg, orally) once a day for twenty-one days. At the end of treatment period, rats were euthanized and eyes were enucleated in Davidson’s solution.
#
Kidney histology
For histological evaluation, kidney samples were embedded in paraffin, and sectioned. These sections (5 µM thickness) were stained with periodic acid–schiff stain. Stained sections were examined using a microscope (DM1000, Leica Microscope) for surface area, glomerulosclerosis and tubulointerstitial fibrosis. Stained sections were examined at 400X magnification by microscope and examined for inflammation. Grading of histopathological findings was done as 0-no abnormalities detected, 1-minimal abnormality (<20%), 2-mild abnormality (21 to 50%), 3-moderate abnormality (51 to 75%), and 4-severe abnormality (76 to 100%) in periodic acid solution (PAS) staining.
#
Immunohistology for C3 deposition in kidney
Deparaffinized kidney sections were processed for antigen retrieval by trypsinization. These sections were washed with TBS-T. Non-specific binding sites were blocked by incubating samples with 5% BSA in TBS for 2 h at 4°C. After washing with TBS-T 2 times, 5 min apart, sections were incubated with anti-C3 antibody (21337-1-AP, 200-folx dilution in 5% BSA in TBS) overnight at 4°C. Unbound antibody was removed by washing the sections with TBS-T. These washed sections were stained with FITC-conjugated secondary antibody to anti-C3 antibody (SA00003-1, 50 X in 5% BSA in TBS) for 1.5 h at 4°C in the dark. The unbound antibody was removed by washing and mounted in a fluoro-shield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI). C3 deposition was observed using in a microscope at 400X magnification (DM1000, Leica Microscope).
#
Eye histology
Eye samples stored in Davidson’s solution for 24 hours were embedded in paraffin, and sectioned. These sections (5 µM thickness) were stained with hematoxylin and eosin (H & E) stain. Stained sections were examined using a microscope (DM1000, Leica Microscope). The thickness of the total retina length, outer nuclear layer and inner nuclear layers were measured. Grading of histopathological findings for retinal folds, degeneration of photoreceptor layers, and disorganization of retinal layers of the retina were performed as 0-no abnormalities detected, 1-minimal abnormality (<20%), 2-mild abnormality (21 to 50%), 3-moderate abnormality (51 to 75%), and 4-severe abnormality (76 to 100%) in H&E staining. Stained sections were examined at 200X magnification by microscope (DM1000, Leica Microscope).
#
Statistical Analysis
Data are presented as mean±standard deviation (SD). All analyses were performed using GraphPad Prism version 10.2 (GraphPad Software, San Diego, CA). Statistical comparisons were conducted by one-way ANOVA, followed by Tukey’s multiple comparison test for group-wise comparisons, except for [Fig. 2d], where intergroup comparison was done with t test. P<0.05 was considered as a statistically significant difference. Histological data were evaluated using Kruskal–Wallis analysis followed by Dunn’s test.
#
#
Results
Desidustat treatment suppressed acute nephritis in mice
Acute nephritis in mice can be induced by drugs or chemicals like LPS, doxorubicin, or BSA, which causes an increase in serum creatinine, serum urea and urinary protein excretion. These symptoms resemble the acute kidney injury observed in humans. LPS (5 mg/kg, IP) treatment in mice induced acute nephritis. In these mice, serum creatinine and urea were significantly increased when compared to saline-treated normal control ([Fig. 1a, b]). Desidustat (15 mg/kg, PO) treatment reduced serum creatinine and urea by 34.1±5.2 and 44.9±5.2%, respectively, when compared to only LPS-treated control mice ([Fig. 1a, b]). Doxorubicin (10 mg/kg, IV) treatment induced glomerulonephritis and demonstrated significant proteinuria, when compared to saline-treated normal control ([Fig. 1c]). Desidustat (15 mg/kg, PO) treatment reduced urinary protein excretion by 47.8±5.8%, when compared to Doxorubicin-treated control mice ([Fig. 1c]). Treatment with BSA caused membranous nephropathy and significantly increased protein excretion in urine when compared to vehicle-treated normal control ([Fig. 1d]). Desidustat (15 mg/kg, PO) reduced urinary excretion of total protein by 40.1±7.6% in BSA-treated mice ([Fig. 1d]). Taken together, these data indicate that desidustat can prevent the symptoms of acute nephritis, glomerulonephritis, and membranous nephropathy in mice.

#
Complement system inhibition by desidustat
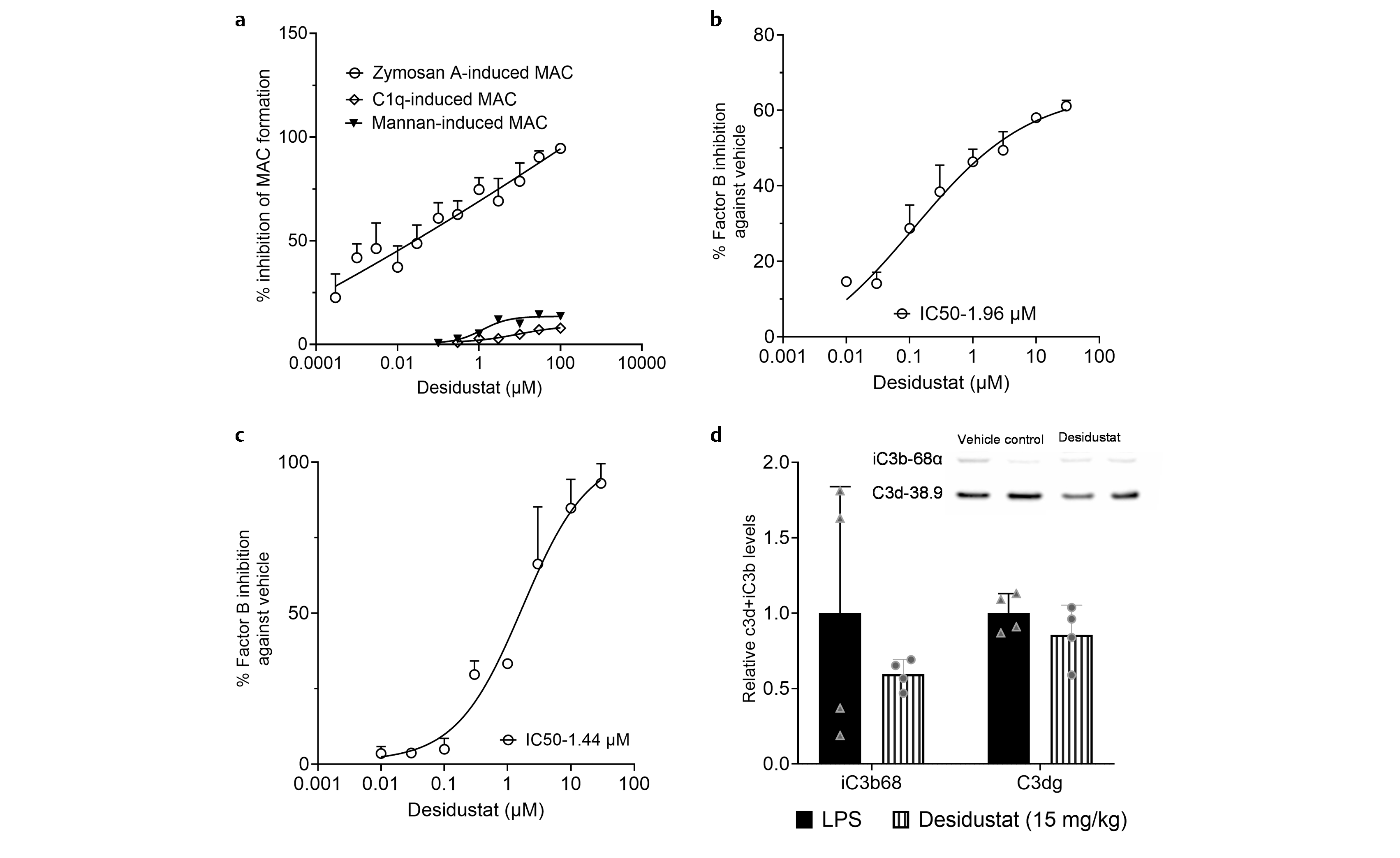
The complement system comprises of alternative, classical, and lectin systems. We investigated the effect of desidustat in these three complement systems. Zymosan A was used for the activation of the alternative complement system, mannan was used for the activation of the lectin complement system, and C1q was used to activate the classical complement system in human serum. Preincubation of desidustat with human serum showed inhibition of the zymosan A-activated complement system in a dose-related manner. The IC50 (Inhibitory concentration, which caused 50% inhibition of MAC -membrane attack complex) was found to be 0.026 µM ([Fig. 2a]). On the other hand, desidustat treatment did not show significant inhibition of either lectin or classical complement pathways ([Fig. 2a]). To investigate whether desidustat can cause a direct inhibition of factor B, the enzymatic conversion of factor B substrate, Ac-SHLGLAR-pNA, to the fluorescent product was estimated, where human or mouse serum was used as the source of factor B. Desidustat treatment exhibited a dose-related inhibition of factor B, with the IC50 of 1.96 and 1.44 µM, in mouse and human serum, respectively ([Fig. 2b, c]). When the complement system in mice was activated with LPS, desidustat treatment inhibited the formation of C3 degradation products, namely iC3b68 and C3dg by 40.5±4.9 and 14.3±9.8%, respectively, when compared to LPS-treated mice ([Fig. 2d]).
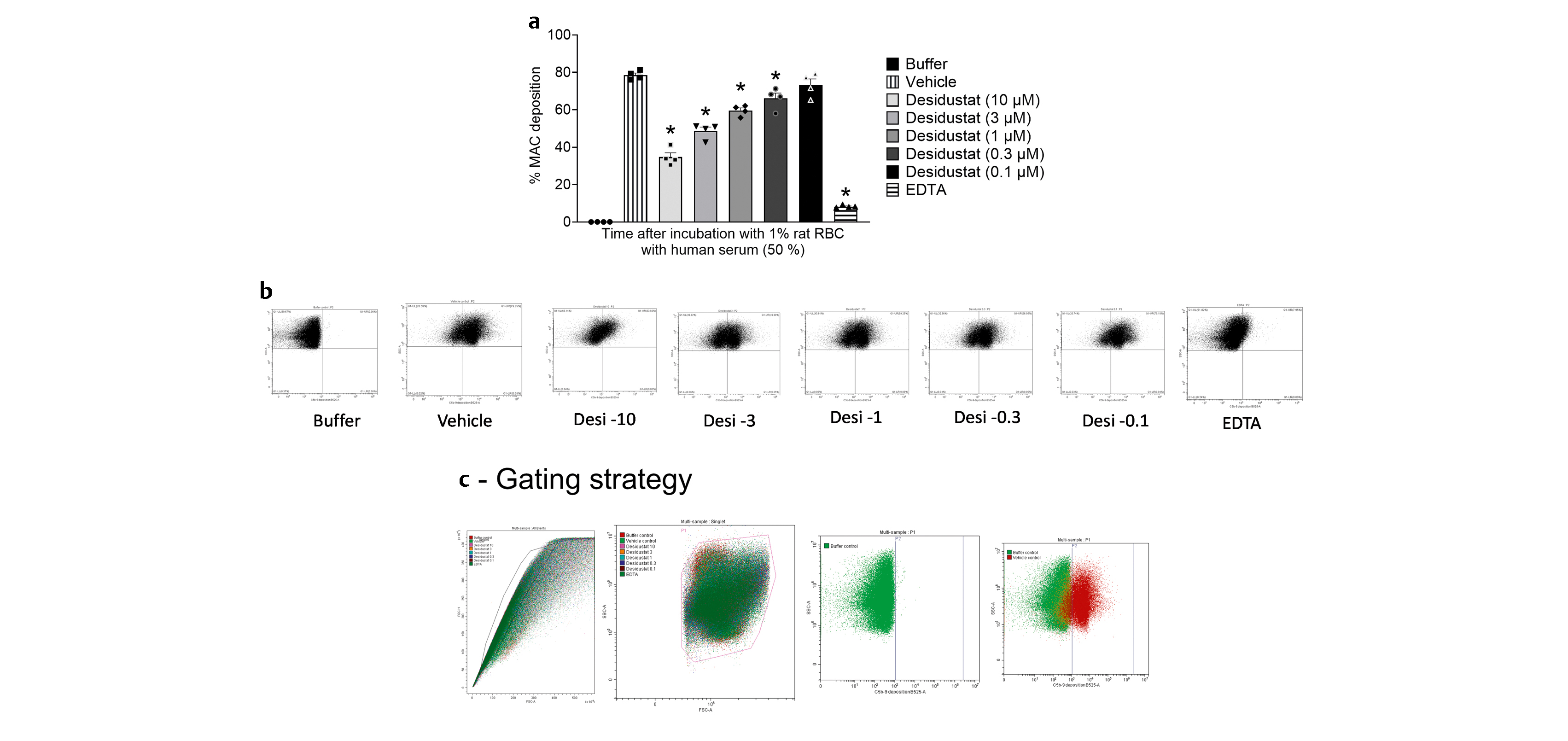
When rat RBCs were incubated with human serum, complement system activation was observed by MAC deposition on the RBC surface, as measured by the flow cytometry. In this experiment, 78.5±1.2% RBC were stained positive for MAC deposition ([Fig. 3a]). The gating strategy used in this analysis was based on scatter plot of forward scatter (FSC)-Area Vs FCS-Height of the singlet cell population. From these cells, MAC positive gate was drawn from unstained buffer control ([Fig. 3b]). Assuming 100% inhibition of alternative complement system in EDTA-treated serum, desidustat at 0.1, 0.3, 1, 3 and 10 µM concentration displayed 7.4±4.5, 17.6±4.0, 26.9±2.0, 42.3±2.9, and 62.3±3.3% inhibition of MAC deposition, respectively, when compared to vehicle control group ([Fig. 3a]). Taken together, these data suggest that desidustat inhibited the alternative complement system, and prevented the generation of C3 degradation products. It appears that this action is achieved, at least partially, by direct inhibition of Factor B.
#
Effect of desidustat on cationic BSA-induced membranous nephropathy
Subcutaneous injections of cationic BSA (cBSA) induce membranous nephropathy in mice. Administration of cBSA is associated with complement system activation which causes symptoms of membranous nephropathy such as proteinuria, increased oxidative stress and inflammation in the kidney. We have observed that cBSA treatment increased the excretion of protein and microalbumin in urine ([Fig. 4a, b]), and also increased malondialdehyde (MDA) and reduced superoxide mutase (SOD) levels in the kidney ([Fig. 4c, d]). Desidustat treatment reduced urinary excretion of protein and microalbumin by 40.4±5.9 and 32.8±3.6%, respectively, compared to cBSA-treated control mice ([Fig. 4a, b]). Oxidative stress markers such as MDA were reduced by 37.0±9.3%, and superoxide mutase activity was increased by 90.8±5.9%, by desidustat treatment, when compared with cBSA-treated control mice ([Fig. 4c, d]).

Interleukin-1ß (IL-1ß) levels in kidney were increased by cBSA-treatment in mice ([Fig. 4e]), along with the thickening of glomerular basement membrane as observed by histopathological assessment ([Fig. 5a]). Desidustat treatment reduced IL-1ß levels in kidney by 60.7±1.2%. It also reduced the thickening of glomerular basement membrane by 24.4±6.9%, when compared with cBSA control ([Fig. 5a, b]). Desidustat treatment also decreased the C3 deposition in the glomerular basement membrane, which was increased by cBSA treatment ([Fig. 5c]). Taken together, in cBSA -induced glomerulonephritis model, desidustat treatment reduced complement deposition in kidney, decreased the symptoms of membranous nephropathy, and also reduced inflammation and oxidative stress.

#
Sodium iodate-induced retinal degeneration
Unwarranted activation of the complement system in the ophthalmic milieu causes retinal degenerative disease. Typically, retinal damage is characterized by the disorganization of retinal layers and the atrophy of retinal cells. In this study, retinal degeneration was induced through intravenous injection of sodium iodate, which decreased the thickness of the outer nuclear layer (ONL) of the retina by 27.5±11.5%, when compared to saline-treated normal control ([Fig. 6a]). Retinal degeneration score as assessed by histopathological screening showed increased retinal folds, degeneration of photoreceptor layers and disorganization of ONL layer by sodium iodate treatment ([Fig. 6b, c]). Desidustat treatment prevented the change in ONL thickness, and also prevented the increase in retinal folds, degeneration of photoreceptor layers and disorganization of ONL layer. Taken together, it appears that desidustat reduced retinal folding and degeneration possibly by inhibition of the complement system.

#
#
Discussion
HIFs are transcription factors that respond to changes in cellular oxygen environment and regulate the expression of various proteins [13]. Inhibition of PHDs prevents degradation of HIFs and increases their stability [4]. Stabilized HIFs regulate erythropoietin (EPO), vascular endothelial growth factor, transforming growth factor-ß, epidermal growth factor, many cytokines and chemokines, along with adhesion molecules that modulate inflammation, cell survival and growth [13]. Additionally, hypoxia also regulates the expression of CD55, a decay-accelerating factor involved in the complement system [14]. Complement system is the key regulator of adaptive immunity and inflammation. HIF-1 is also linked to active-C3, a key mediator of complement activation [15].
Inflammatory stimuli such as lipopolysaccharide (LPS) and bovine serum albumin (BSA) activate the complement system [16] [17]. Apart from these, doxorubicin can also induce kidney damage by complement system activation [18]. In this study, we have used desidustat a known PHD inhibitor and stabilizer of HIF [19], to assess the inhibitory potential using these in vivo models. Desidustat reduced serum creatinine and urea in LPS-induced acute kidney injury. Proteinuria is an important diagnostic marker for glomerulonephritis associated with complement system dysregulation [20]. Desidustat treatment also reduced proteinuria in doxorubicin and BSA overload-induced glomerulonephritis.
There are three major pathways of complement system activation namely; classic, lectin and alternative complement pathways. Of these three, the alternative complement pathway is the major pathway that amplifies and converges the other two pathways [21]. Zymosan A activates an alternative complement system [22]. Membrane attack complex formation (MAC or c5b-9) was reduced by desidustat treatment in a dose-related manner in the zymosan A activated complement system, while lectin and classic complement pathways were not affected. The alternative complement system pathway can be inhibited at C3, C5, C9, properdin, factor H, factor B, factor D, CD55, and CD59, but factor B is of greater importance in regulating alternative complement system [22]. Desidustat inhibited factor B (human as well as mice) in a dose-related manner. Hemolysis of rat RBCs using human serum as a source of complement factors was employed in the assay for complement-mediated hemolysis in this study, wherein, desidustat treatment reduced the deposition of MAC on the rat RBC surface in a dose-related manner. Thus, it appears that desidustat can reduce the MAC inhibition at least partially by inhibition of factor B. In vivo inhibition of factor B can be assessed by estimation of inhibition of the C3 degradation products because C3 convertase and factor B cleave C3/C3(H2O) into C3a, C3b, and ic3d/g. We have observed that desidustat treatment could inhibit the C3 degradation that was induced by the LPS challenge in mice. Roxadustat, a HIF stabilizer, suppresses C1q levels, a protein involved in the activation of classical complement system, and thus may prevent classical complement activation [23]. Since the HIF-stabilizer therapy is required to be given chronically to anemia patients, changes in complement pathways, especially classical or lectin pathways, may pose a toxicity challenge. We have investigated the effect of desidustat on the activation of classical and lectin pathways. It appears that desidustat modulates only the alternative pathway, without affecting the classical and lectin-mediated complement pathways. Though further clinical studies directed at the complement-mediated effects in humans would be needed to ascertain this preclinical finding, it appears that desidustat specifically inhibits the alternative complement pathway, without affecting the classical or lectin pathways. We have also observed the anti-inflammatory effects of desidustat in cBSA-induced glomerulonephritis in mice. Coupled with the previously reported anti-inflammatory actions of desidustat, desidustat may not cause the toxicity associated with a decrease in classical or lectin-mediated complement activation [6] [8].
Membranous nephropathy (MN) is a major cause of idiopathic nephrotic syndrome, which may progress to end-stage kidney disease [24]. Podocyte injury, MAC deposition, and, the expansion of the glomerular basement membrane cause proteinuria and induce the nephrotic syndrome. Exogenous cationic proteins, such as cationic BSA (cBSA), bind to anionic sites in the glomerular basement membrane (GBM) barrier and form subepithelial immune complex recapitulating membranous nephropathy mimicking clinical characteristics of nephrotic syndrome [24]. Membranous nephropathy models do not show elevated blood urea nitrogen and serum creatinine, while diffuse thickening of the glomeruli basement membrane without hypercellularity is always evident in histology, with increased deposition of C3 [24]. Desidustat treatment reduced C3 deposition in kidney, and also reduced proteinuria and microalbuminuria in cBSA-induced membranous nephropathy in mice. Proteinuria and microalbuminuria are associated with increased lipid peroxidation in the kidney [25]. Reduced activity of superoxide dismutase (SOD) in glomerular suppresses scavenging reaction increasing the susceptibility of glomeruli to oxidative stress [25]. In the current study, desidustat treatment reduced MDA and increased SOD activity in the kidney, indicating an increased scavenging effect and reduced oxidative stress. Complement activation causes the recruitment of inflammatory cells and releases cytokines and chemokines [26]. Interleukin-1 is a cytokine released after the recruitment of inflammatory cells which further increases the synthesis of complement components and factor B [27]. In the present study, cBSA-induced cytokine release was reduced by desidustat treatment, and it also reduced GBM thickening, suggesting a beneficial role of desidustat, a clinically used PHD inhibitor for treatment of complement-mediated kidney disease.
Age-related macular degeneration (AMD) is a chronic and progressive degenerative retinal disease. Overactivation of the alternative complement pathway is one of the main pathogenic factors for the progression of AMD [28]. Sodium iodate induces retinal degeneration and causes a decrease in visual activity [29], by activation of the complement system and increasing the deposition of complement protein in the retina [30]. Desidustat treatment prevented the decrease in ONL thickness, number of retinal folds and disorganization of ONL, which suggests the protective effect of PHD inhibition on retinal degeneration.
Several HIF stabilizers, such as roxadustat, daprodustat, vadadustat, molidustat, and enarodustat, are used in the treatment of anemia of CKD. All these PHD inhibitors show nanomolar potency in PHD binding assays, whereas daprodustat shows a suboptimal effect (Emax) in HIF stabilization assay, which may have been translated in lesser EPO release caused by daprodustat in preclinical as well as clinical studies [31]. On the other hand, the reticulocyte count, which is the hallmark of erythropoiesis, was found to be significantly increased with desidustat, as compared to vadadustat. PHD inhibitors have been reported to have toxicities related to accelerated pharmacology, arising out of elevated haemoglobin. However, no off-target toxicities were observed following desidustat treatment, as against more cancer-related death or tumor progression events and more esophageal or gastric erosion events in the daprodustat treatment [32] [33]. Roxadustat trial indicated a higher portion of non dialysis-dependent chronic kidney disease patients and dialysis-dependent chronic kidney disease patients experienced vascular access thrombosis compared with controls [34]. Compared to other PHD inhibitors, desidustat treatment, either in nondialysis-dependent patients or in dialysis-dependent patients is noninferior to either epoetin alpha or darbepoetin, without any serious side effect related to tumor progression, gastrointestinal trouble, or thrombosis [32]. Desidustat treatment also showed a prominent decrease in hepcidin levels [35]. Taken together, desidustat treatment is found to be effective and safe in CKD-induced anemia patients.
This study suggests that PHD inhibitor desidustat reduces aberrant activation of the alternative complement system, partially through factor B inhibition. Desidustat treatment also reduced inflammation and oxidative stress and prevented cBSA-induced glomerulonephritis (membranous nephropathy) and sodium iodate-induced retinal degeneration in preclinical models. Thus, in addition to its established role in the treatment of CKD-induced anemia, desidustat can be a potentially useful therapy for complement-mediated diseases.
#
#
Conflict of Interest
All the authors are employees of Zydus Research Centre, Zydus Lifesciences Limited, Ahmedabad, India. No author of this manuscript has any potential competing interest.
Acknowledgement
The authors are grateful to Mr. Pankaj R. Patel, Chairman, Zydus Lifesciences Limited, for his guidance and support.
-
References
- 1 Sjöberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol 2009; 30: 83-90
- 2 Poppelaars F, Thurman JM. Complement-mediated kidney diseases. Mol Immunol 2020; 128: 175-187
- 3 Colgan SP, Furuta GT, Taylor CT. Hypoxia and Innate Immunity: Keeping Up with the HIFsters. Annu Rev Immunol 2020; 38: 341-363
- 4 Joharapurkar AA, Pandya VB, Patel VJ. et al. Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases. J Med Chem 2018; 61: 6964-6982
- 5 Patel V, Joharapurkar A, Kshirsagar S. et al. Hepcidin inhibition improves iron homeostasis in ferrous sulfate and LPS treatment model in mice. Drug Res 2021; 71: 528-534
- 6 Jain M, Joharapurkar A, Patel V. et al. Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation. Eur J Pharmacol 2019; 843: 113-120
- 7 Parmar DV, Kansagra KA, Patel JC. et al. on behalf of the ZYAN1 Trial Investigators. Outcomes of Desidustat Treatment in People with Anemia and Chronic Kidney Disease: A Phase 2 Study. Am J Nephrol 2019; 49: 470-478
- 8 Joharapurkar AA, Patel VJ, Kshirsagar SG. et al. Prolyl hydroxylase inhibitor desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines and oxidative stress. Drug Dev Res 2021; 82: 852-860
- 9 Animals NRC (US) C for the U of the G for the, Laboratory C and U of. Guide for the Care and Use of Laboratory Animals. Washington, D.C.: National Academies Press; 2011
- 10 Harboe M, Garred P, Lindstad JK. et al. The Role of Properdin in Zymosan- and Escherichia coli-Induced Complement Activation. The Journal of Immunology 2012; 189: 2606-2613
- 11 Le GT, Abbenante G, Fairlie DP. Profiling the enzymatic properties and inhibition of human complement factor B. J Biol Chem 2007; 282: 34809-34816
- 12 Wang W, Sheng L, Chen Y. et al. Total coumarin derivates from Hydrangea paniculata attenuate renal injuries in cationized-BSA induced membranous nephropathy by inhibiting complement activation and interleukin 10-mediated interstitial fibrosis. Phytomedicine 2022; 96: 153886
- 13 Patel VJ, Joharapurkar A, Jain MR. The Perspective of Using Flow Cytometry for Unpuzzling Hypoxia-Inducible Factors Signalling. Drug Res 2023; 74: 113-122
- 14 Pandya PH, Fisher AJ, Mickler EA. et al. Hypoxia-Inducible Factor-1α Regulates CD55 in Airway Epithelium. Am J Respir Cell Mol Biol 2016; 55: 889-898
- 15 Khan MA, Shamma T, Kazmi S. et al. Hypoxia-induced complement dysregulation is associated with microvascular impairments in mouse tracheal transplants. J Transl Med 2020; 18: 147
- 16 Fu X, Ju J, Lin Z. et al. Target deletion of complement component 9 attenuates antibody-mediated hemolysis and lipopolysaccharide (LPS)-induced acute shock in mice. Sci Rep 2016; 6: 30239
- 17 Ikeda Y, Horinouchi Y, Hamano H. et al. Dietary iron restriction alleviates renal tubulointerstitial injury induced by protein overload in mice. Sci Rep 2017; 7: 10621
- 18 Tang Z, Lu B, Hatch E. et al. C3a Mediates Epithelial-to-Mesenchymal Transition in Proteinuric Nephropathy. Journal of the American Society of Nephrology 2009; 20: 593-603
- 19 Jain MR, Joharapurkar AA, Pandya V. et al. Pharmacological Characterization of ZYAN1, a Novel Prolyl Hydroxylase Inhibitor for the Treatment of Anemia. Drug Res 2016; 66: 107-112
- 20 Madaio MP, Harrington JT. The Diagnosis of Glomerular Diseases: acute glomerulonephritis and the nephrotic syndrome. Arch Intern Med 2001; 161: 25-34
- 21 Janeway C, Travers P, Walport M. et al. Immunobiology: The Immune System in Health and Disease. 5. Aufl. New York: Garland Science; 2001
- 22 Schubart A, Anderson K, Mainolfi N. et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proceedings of the National Academy of Sciences 2019; 116: 7926-7931
- 23 Kiriakidis S, Hoer SS, Burrows N. et al. Complement C1q is hydroxylated by collagen prolyl 4 hydroxylase and is sensitive to off-target inhibition by prolyl hydroxylase domain inhibitors that stabilize hypoxia-inducible factor. Kidney Int 2017; 92: 900-908
- 24 Borza DB, Zhang JJ, Beck LH. et al. Mouse models of membranous nephropathy: the road less travelled by. Am J Clin Exp Immunol 2013; 2: 135-145
- 25 Motiram Kakalij R, Tejaswini G, Patil MA. et al. Vanillic Acid Ameliorates Cationic Bovine Serum Albumin Induced Immune Complex Glomerulonephritis in BALB/c Mice. Drug Dev Res 2016; 77: 171-179
- 26 Ostrycharz E, Hukowska-Szematowicz B. New Insights into the Role of the Complement System in Human Viral Diseases. Biomolecules 2022; 12: 226
- 27 Dinarello C. Interleukin-1 and interleukin-1 antagonism. Blood 1991; 77: 1627-1652
- 28 Armento A, Ueffing M, Clark SJ. The complement system in age-related macular degeneration. Cellular and Molecular Life Sciences 2021; 78: 4487-4505
- 29 Chowers G, Cohen M, Marks-Ohana D. et al. Course of sodium iodate–induced retinal degeneration in albino and pigmented mice. Invest Ophthalmol Vis Sci 2017; 58: 2239-2249
- 30 Wang S, Du L, Yuan S. et al. Complement C3a receptor inactivation attenuates retinal degeneration induced by oxidative damage. Front Neurosci 2022; 16: 951491
- 31 Janssens LK, Stove CP. Sensing an Oxygen Sensor: Development and Application of Activity-Based Assays Directly Monitoring HIF Heterodimerization. Anal Chem 2021; 93: 14462-14470
- 32 Gang S, Khetan P, Varade D. et al. Study Investigator Group. Desidustat in Anemia due to Dialysis-Dependent Chronic Kidney Disease: A Phase 3 Study (DREAM-D). Am J Nephrol 2022; 53: 343-351
- 33 Johnson HN, Prasad-Reddy L. Daprodustat: A Hypoxia-Inducible Factor-Prolyl Hydroxylase Inhibitor for Anemia of Chronic Kidney Disease. Ann Pharmacother 2024; 14 10600280241241563 Online ahead of print
- 34 McCallum W, Weiner DE. HIF-PHIs for Anemia Management in CKD: Potential and Uncertainty ASCEND. Clin J Am Soc Nephrol 2022; 17: 1255-1258
- 35 Kansagra KA, Parmar D, Jani RH. et al. Phase I Clinical Study of ZYAN1, A Novel Prolyl-Hydroxylase (PHD) Inhibitor to Evaluate the Safety, Tolerability, and Pharmacokinetics Following Oral Administration in Healthy Volunteers. Clin Pharmacokinet 2018; 57: 87-102
Correspondence
Publikationsverlauf
Eingereicht: 26. März 2024
Angenommen: 17. Juni 2024
Artikel online veröffentlicht:
11. Juli 2024
© 2024. Thieme. All rights reserved.
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Sjöberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol 2009; 30: 83-90
- 2 Poppelaars F, Thurman JM. Complement-mediated kidney diseases. Mol Immunol 2020; 128: 175-187
- 3 Colgan SP, Furuta GT, Taylor CT. Hypoxia and Innate Immunity: Keeping Up with the HIFsters. Annu Rev Immunol 2020; 38: 341-363
- 4 Joharapurkar AA, Pandya VB, Patel VJ. et al. Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases. J Med Chem 2018; 61: 6964-6982
- 5 Patel V, Joharapurkar A, Kshirsagar S. et al. Hepcidin inhibition improves iron homeostasis in ferrous sulfate and LPS treatment model in mice. Drug Res 2021; 71: 528-534
- 6 Jain M, Joharapurkar A, Patel V. et al. Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation. Eur J Pharmacol 2019; 843: 113-120
- 7 Parmar DV, Kansagra KA, Patel JC. et al. on behalf of the ZYAN1 Trial Investigators. Outcomes of Desidustat Treatment in People with Anemia and Chronic Kidney Disease: A Phase 2 Study. Am J Nephrol 2019; 49: 470-478
- 8 Joharapurkar AA, Patel VJ, Kshirsagar SG. et al. Prolyl hydroxylase inhibitor desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines and oxidative stress. Drug Dev Res 2021; 82: 852-860
- 9 Animals NRC (US) C for the U of the G for the, Laboratory C and U of. Guide for the Care and Use of Laboratory Animals. Washington, D.C.: National Academies Press; 2011
- 10 Harboe M, Garred P, Lindstad JK. et al. The Role of Properdin in Zymosan- and Escherichia coli-Induced Complement Activation. The Journal of Immunology 2012; 189: 2606-2613
- 11 Le GT, Abbenante G, Fairlie DP. Profiling the enzymatic properties and inhibition of human complement factor B. J Biol Chem 2007; 282: 34809-34816
- 12 Wang W, Sheng L, Chen Y. et al. Total coumarin derivates from Hydrangea paniculata attenuate renal injuries in cationized-BSA induced membranous nephropathy by inhibiting complement activation and interleukin 10-mediated interstitial fibrosis. Phytomedicine 2022; 96: 153886
- 13 Patel VJ, Joharapurkar A, Jain MR. The Perspective of Using Flow Cytometry for Unpuzzling Hypoxia-Inducible Factors Signalling. Drug Res 2023; 74: 113-122
- 14 Pandya PH, Fisher AJ, Mickler EA. et al. Hypoxia-Inducible Factor-1α Regulates CD55 in Airway Epithelium. Am J Respir Cell Mol Biol 2016; 55: 889-898
- 15 Khan MA, Shamma T, Kazmi S. et al. Hypoxia-induced complement dysregulation is associated with microvascular impairments in mouse tracheal transplants. J Transl Med 2020; 18: 147
- 16 Fu X, Ju J, Lin Z. et al. Target deletion of complement component 9 attenuates antibody-mediated hemolysis and lipopolysaccharide (LPS)-induced acute shock in mice. Sci Rep 2016; 6: 30239
- 17 Ikeda Y, Horinouchi Y, Hamano H. et al. Dietary iron restriction alleviates renal tubulointerstitial injury induced by protein overload in mice. Sci Rep 2017; 7: 10621
- 18 Tang Z, Lu B, Hatch E. et al. C3a Mediates Epithelial-to-Mesenchymal Transition in Proteinuric Nephropathy. Journal of the American Society of Nephrology 2009; 20: 593-603
- 19 Jain MR, Joharapurkar AA, Pandya V. et al. Pharmacological Characterization of ZYAN1, a Novel Prolyl Hydroxylase Inhibitor for the Treatment of Anemia. Drug Res 2016; 66: 107-112
- 20 Madaio MP, Harrington JT. The Diagnosis of Glomerular Diseases: acute glomerulonephritis and the nephrotic syndrome. Arch Intern Med 2001; 161: 25-34
- 21 Janeway C, Travers P, Walport M. et al. Immunobiology: The Immune System in Health and Disease. 5. Aufl. New York: Garland Science; 2001
- 22 Schubart A, Anderson K, Mainolfi N. et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proceedings of the National Academy of Sciences 2019; 116: 7926-7931
- 23 Kiriakidis S, Hoer SS, Burrows N. et al. Complement C1q is hydroxylated by collagen prolyl 4 hydroxylase and is sensitive to off-target inhibition by prolyl hydroxylase domain inhibitors that stabilize hypoxia-inducible factor. Kidney Int 2017; 92: 900-908
- 24 Borza DB, Zhang JJ, Beck LH. et al. Mouse models of membranous nephropathy: the road less travelled by. Am J Clin Exp Immunol 2013; 2: 135-145
- 25 Motiram Kakalij R, Tejaswini G, Patil MA. et al. Vanillic Acid Ameliorates Cationic Bovine Serum Albumin Induced Immune Complex Glomerulonephritis in BALB/c Mice. Drug Dev Res 2016; 77: 171-179
- 26 Ostrycharz E, Hukowska-Szematowicz B. New Insights into the Role of the Complement System in Human Viral Diseases. Biomolecules 2022; 12: 226
- 27 Dinarello C. Interleukin-1 and interleukin-1 antagonism. Blood 1991; 77: 1627-1652
- 28 Armento A, Ueffing M, Clark SJ. The complement system in age-related macular degeneration. Cellular and Molecular Life Sciences 2021; 78: 4487-4505
- 29 Chowers G, Cohen M, Marks-Ohana D. et al. Course of sodium iodate–induced retinal degeneration in albino and pigmented mice. Invest Ophthalmol Vis Sci 2017; 58: 2239-2249
- 30 Wang S, Du L, Yuan S. et al. Complement C3a receptor inactivation attenuates retinal degeneration induced by oxidative damage. Front Neurosci 2022; 16: 951491
- 31 Janssens LK, Stove CP. Sensing an Oxygen Sensor: Development and Application of Activity-Based Assays Directly Monitoring HIF Heterodimerization. Anal Chem 2021; 93: 14462-14470
- 32 Gang S, Khetan P, Varade D. et al. Study Investigator Group. Desidustat in Anemia due to Dialysis-Dependent Chronic Kidney Disease: A Phase 3 Study (DREAM-D). Am J Nephrol 2022; 53: 343-351
- 33 Johnson HN, Prasad-Reddy L. Daprodustat: A Hypoxia-Inducible Factor-Prolyl Hydroxylase Inhibitor for Anemia of Chronic Kidney Disease. Ann Pharmacother 2024; 14 10600280241241563 Online ahead of print
- 34 McCallum W, Weiner DE. HIF-PHIs for Anemia Management in CKD: Potential and Uncertainty ASCEND. Clin J Am Soc Nephrol 2022; 17: 1255-1258
- 35 Kansagra KA, Parmar D, Jani RH. et al. Phase I Clinical Study of ZYAN1, A Novel Prolyl-Hydroxylase (PHD) Inhibitor to Evaluate the Safety, Tolerability, and Pharmacokinetics Following Oral Administration in Healthy Volunteers. Clin Pharmacokinet 2018; 57: 87-102