

Subscribe to RSS
DOI: 10.1055/a-2766-4857
Light-Driven Stereoselective Aryl Bisenone Cycloaddition: A Metal-Free Strategy for the Construction of Functionalized Pyrrolidines and Tetrahydrofurans
Authors
This work was supported by: (a) MUSA - Multilayered Urban Sustainability Action – project, funded by the European Union – NextGenerationEU, under the National Recovery and Resilience Plan (NRRP) Mission 4 Component 2 Investment Line 1.5: Strengthening of research structures and creation of R&D ‘innovation ecosystems’, set up of ‘territorial leaders in R&D’; (b) PRIN 2022 grant ‘Enabling technologies for sustainable and innovative catalytic transformations - BEST-CAT’ (CUP G53D23003260006); (c) PSR 2025 grant ‘Catalytic approaches to the sustainable synthesis of high added-value fine chemicals’ by the Università degli Studi di Milano.
Abstract
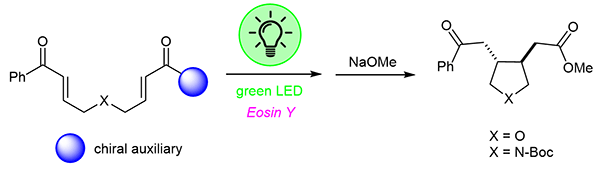
In this study, we report an exploratory study on the reductive cyclization of nitrogen- and oxygen-containing aryl bisenones, enabling the efficient synthesis of highly functionalized pyrrolidines and tetrahydrofurans under visible light irradiation. The transformation proceeds through a photocatalytic cyclization promoted by catalytic amounts of Eosin Y, with Schreiner’s thiourea and Hantzsch ester acting as mediators under visible green light irradiation. The transformation is stereoselective and affords the five-membered heterocycle as a single, trans isomer. Moreover, the introduction of chiral oxazolidinone auxiliaries onto aryl bisenone precursors allowed the formation of chiral, highly substituted trans pyrrolidines and tetrahydrofurans in good yields, although with modest stereoselectivities.
Key words
photocatalysis - photocatalytic cyclization - Eosin Y - stereoselective synthesis - aryl bis-enones - pyrrolidines - tetrahydrofuransIn recent years, visible light transformations promoted by organic dyes have emerged as a powerful and sustainable tool for the synthesis of highly functionalized molecular frameworks.[1]
Among the various light-driven reactions, photocatalytic cyclization of bisenone derivatives have proven to be a versatile strategy for the synthesis of highly functionalized cyclopentanes and bicyclo[3.2.0]heptanes. Starting from the same bisenone precursor, monocyclic alkanes are formed in the presence of a Brønsted acid in protic media, whereas Lewis acids in aprotic solvents promote the formation of bicyclic products. This class of transformations, initially explored through cobalt-catalyzed anion-radical cycloadditions by Krische and co-workers,[2] was subsequently extended by exploiting radical homogeneous electron transfer strategies[3] and then visible light activation, as reported by Yoon[4] and Zeitler and co-workers.[5] More recently, the use of chiral auxiliaries, such as Evans’ oxazolidinones, has enabled the development of a stereoselective, visible-light-catalyzed reductive cyclization of aryl enones for the synthesis of enantiomerically enriched trans-1,2-cyclopentanes[6] and chiral bicyclo[3.2.0]heptanes;[7] [8] these reactions have been further investigated through the implementation of continuous-flow methodologies and the use of photocatalysts supported on silica[9] and polymeric materials.[8]
Many different transition metal complexes or organic dyes, have been successfully employed as photocatalysts for the reductive photocyclization of bisenones. Among them, Eosin Y[10] has emerged as a particularly efficient, metal-free catalyst capable of promoting these transformations under mild reaction conditions.[5] [6] [7] [8] [9] Inspired by these strategies reported in the literature for the synthesis of 1,2-substituted cyclopentanes, and with the aim of expanding the photocycloaddition to heterocyclic targets, we envisioned the possibility to extend the photocatalytic cyclization of bisenones to nitrogen- and oxygen-containing analogues, enabling the formation of pyrrolidines and tetrahydrofurans.
Different synthetic strategies were explored for the synthesis of symmetric oxygen- and nitrogen-containing aryl bisenones 7 and 8; among these approaches, the most efficient results were obtained following the known procedure outlined in Scheme [1]. The synthesis of compounds 7 and 8 features ylide 2 as common intermediate.

Ylide 2 was obtained in almost quantitative yield by reacting α-bromoacetophenone (1) with triphenylphosphine at reflux for 24 h followed by a deprotonation with 2 M NaOH solution, according a standard Wittig protocol.[11] Commercially available 2,5-dihydrofuran (3) and N-Boc-protected 2,5-dihydro-1H-pyrrole 4 were subjected to ozonolysis and afforded the corresponding aldehydes 5 and 6 which were not isolated but directly reacted in situ with the previously formed ylide 2, yielding the desired bisenones 7 and 8 in 48% and 29% yield, respectively, after chromatographic purification. Having synthesized oxygen- and nitrogen-containing aryl bisenones 7 and 8, we next investigated their behavior in the green-light-promoted cycloaddition (Scheme [2]).

Starting from the protocol developed by Zeitler,[5] and after a brief screening of the reaction conditions, it was found that, in the presence of 2.5 mol% of Eosin Y, 20 mol% of Schreiner’s thiourea, and 1.1 equiv. of di-tert-butyl 1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (tBu-Hantzsch ester) and 1 equiv. of N,N-diisopropylethylamine (iPr2NEt) in degassed dichloromethane at room temperature under green light irradiation (green LEDs, λ = 530 nm, I = 424 mW/cm2), symmetric aryl bisenone 7 underwent efficient intramolecular photocycloaddition to afford trans-tetrahydrofuran 9 in 51% isolated yield after chromatographic purification. Applying the same photocatalytic protocol to compound 8 resulted in a remarkably faster transformation, delivering substituted trans-pyrrolidine 10 in 89% yield after 4 hours of irradiation.
The exclusive formation of the trans products is in agreement with the stereochemical outcome generally observed in intramolecular photocycloaddition of bisenones.[4] , [6] [7] [8]
Having established the feasibility of synthesizing pyrrolidines and tetrahydrofurans by Eosin Y photocatalyzed reaction, the cyclization was further explored by examining a series of chiral nitrogen- and oxygen-containing aryl bisenones. Evans’ oxazolidinones were incorporated into the bisenone backbone, since this strategy has been previously demonstrated to provide effective stereocontrol in photocycloadditions.[6] [7]
Initially, bisenones 15a–c and 16, bearing different chiral oxazolidinones, were synthesized following the procedure outlined in Scheme [3]. The treatment of oxazolidinones 11a–c with 2-chloroacetyl chloride afforded compounds 12a–c in 78%, 87%, and 51% yield, respectively. These intermediates were converted into the corresponding ylides 13a–c by reaction with PPh3 in DCM for 48 hours followed by quenching with aqueous NaOH. Compound 14 was synthesized via a three-step sequence starting from the N-Boc-protected 2,5-dihydro-1H-pyrrole 4, that was first subjected to ozonolysis (O3, 0.7 bar) in DCM at –78 °C, to afford the ozonide that was reduced with dimethyl sulfide and treated with 1 equiv. of ylide 2 to afford the target compound in 35% yield, with selective monosubstitution achieved by careful control of the stoichiometric ratios (see the Supporting Information). Compound 14 was then reacted with the previously synthesized 13a–c derivatives, to afford enantiopure bisenones 15a–c by treatment with MgSO4 in DCM for 16 hours. Compounds 15a–c were isolated in 31%, 23%, and 48% yield, respectively, with the (2E,7E) diastereoisomer as the major product, which was easily separated through chromatographic purification. By analogy, compound 17 was obtained by reacting compound 13c with aldehyde 16 which was synthesized from 2,5-dihydrofuran 3 according to a literature procedure.[4]

The photocyclization of bisenone derivatives 15a–c and 17 was explored under visible light irradiation using Eosin Y as a photosensitizer according to the standard reaction conditions (2.5 mol% of Eosin Y, 20 mol% of Schreiner’s thiourea, 1.1 equiv. of tBu-Hantzsch ester, and 1 equiv. of iPr2NEt, room temperature, green light). Cycloaddition of 15a–c leads to the formation of products 18a–c in modest to good yields. However, the diastereomeric ratio could not be directly determined from the 1H NMR spectrum of the crude reaction mixture due to signals overlapping in the aliphatic region, which prevent reliable integration. For this reason, compounds 18a–c were converted into the corresponding methyl ester 20, obtained after removal of the chiral auxiliary by treatment with sodium methoxide in dry methanol at –20 °C for 18 hours. The resulting ester was then analyzed by HPLC on a chiral stationary phase, enabling accurate assessment of the enantiomeric excess of the product (see Supporting Information for further details). The results are reported in Table [1]. Starting from bisenone 15a, methyl ester 20 was recovered in 53% overall yield as a racemic mixture (entry 1). Starting from bisenone 15b, methyl ester 20 was recovered in 80% overall yield and 55:45 er, while the more sterically hindered bisenone 15c afforded product 20 in 42% yield, with enhanced selectivity (er = 67:33), indicating that bulkier substituents favor the formation of one preferred isomer. In the case of the oxygen-containing bisenone 17, substituted tetrahydrofuran 21 was obtained in 43% overall yield, as single trans isomer and very poor stereoselectivity (58:42, entry 4). Lower yields and no significant change in the ee were observed when the reaction was run at lower temperature (–20 °C), probably due to the long distance of the chiral auxiliary from the reacting functional groups, which make difficult, and not efficient, the control of the stereochemical outcome.
a Enantiomeric ratio (er) determined on methyl esters 20 and 21 after the removal of oxazolidinone moiety by treatment of 18a–c and 19 with NaOMe in dry MeOH for 18 h at –20 °C. See Supporting Information for further details.
All products were obtained in trans configuration.[4] According to the literature,[4] [5] [6] [7] a proposed mechanism for this kind of transformation is reported in Scheme [4]. After irradiation with visible green light, the excited state of Eosin Y undergoes a SET reductive quenching with iPr2NEt or Hantzsch’s ester to generate the corresponding Eosin Y radical anion and the amino radical cation. The radical anion of Eosin Y in its photoexcited state is a strong reducing agent and can promote a SET with the unsaturated aryl ketone A activated through hydrogen by the thiourea, thus restoring the photocatalyst. The resulting 1,4-distonic radical anion B undergoes a cycloaddition to afford the cycloalkyl ketyl radical C, that is trapped by direct hydrogen transfer from the radical cation of the reductive quencher to release the product D in a trans-conformation.

According to this proposed mechanism, it can be assumed that the hydrogen bond donor moiety of the thiourea plays a dual role: activating the bisenone toward SET through hydrogen bonding and, at the same way, promoting a proton transfer to selectively afford the cyclic product.[5]
In conclusion, this study demonstrates that visible-light-mediated photocyclization of nitrogen- and oxygen-containing aryl bisenones constitutes a practical strategy to access functionalized chiral pyrrolidines and tetrahydrofurans. The compounds could be obtained under mild, metal-free conditions. in moderate to good yields, as single trans isomer, although with poor control of the absolute stereochemistry.
1H NMR spectra were recorded at 300 MHz (Brucker AV300) using the solvent reference relative to TMS. 13C NMR spectra were recorded on a 300 MHz spectrometer (Bruker AV300) operating at 75 MHz, with complete proton decoupling, relative to TMS with the respective solvent resonance as the internal standard (CDCl3: δ = 77.0). 31P NMR spectra were recorded on a 300 MHz spectrometer (Bruker AV300) operating at 122 MHz, with complete proton decoupling, relative to 85% H3PO4 in water (δ = 0). Mass spectra were registered on an APEX II & Xmass software (Bruker Daltonics) instrument or on a Thermo Finnigan LCQ Advantage instrument, equipped with an APCI ion source. HPLC analyses were conducted on a chiral stationary phase, using a CHIRALCEL OJ-H column to determine enantiomeric excess, using an Agilent Instrument Series 1100. The specific operative conditions for each product are reported in the corresponding section. The purification of products was performed by column chromatography with flash technique (according to the Still method) using as stationary phase silica gel 230–400 mesh purchased from Sigma Aldrich. Reactions and chromatographic purifications were monitored by analytical TLC using silica gel 60 F254 pre-coated glass plates and visualized using UV light (365 nm) or KMnO4 solution.
1-Phenyl-2-(triphenylphosphanylidene)ethanone (2)
In a round-bottom flask, PPh3 (1.343 g, 5.1 mmol, 1 equiv.) was dissolved in dry DCM (5 mL) under N2 atmosphere. A previously prepared solution of α-bromoacetophenone (992 mg, 5 mmol, 1 equiv.) in dry DCM (10 mL) was added, and the mixture was stirred for 24 h at r.t. The crude was evaporated in vacuo and washed with Et2O (3 × 15 mL). The phosphonium salt was obtained pure as a white powder. [NOTE: the phosphonium salt is stable and can be stored at r.t.]
The phosphonium salt was dissolved in DCM (10 mL) and then 2 M NaOH solution (10 mL) was slowly poured into the mixture under vigorous stirring. The mixture was stirred vigorously for 3 h, then the organic phase was separated, washed with brine (3 × 10 mL), dried (Na2SO4), filtered, and concentrated in vacuo. The desired product 2 was obtained as a foaming sticky oil (1.7 g, 4.3 mmol, 86%), in agreement with literature data.[6]
1H NMR (300 MHz, CDCl3): δ = 8.00 (d, J = 3.4 Hz, 2 H), 7.82–7.65 (m, 4 H), 7.65–7.41 (m, 6 H), 7.41–7.32 (m, 2 H), 7.28 (d, J = 1.4 Hz, 2 H), 4.48 (s, 2 H), 4.41 (s, 3 H).
31P NMR (122 MHz, CDCl3): δ = 16.60 (s).
Oxazolidinones 11a–c; General Procedure
A 100-mL round-bottom flask, equipped with a Vigreux column and a magnetic stirring bar, was charged with the desired α-amino alcohol (1 equiv.), diethyl carbonate (2 equiv.), and K2CO3 (1 equiv.). The mixture was stirred and heated to 135 °C until EtOH was completely distilled off (approx. 1–2 h). After completion, the mixture was cooled to r.t., diluted with water (40 mL), and extracted with DCM (3 × 30 mL). The combined organic phases were washed with brine (40 mL), dried (anhyd Na2SO4), filtered, and concentrated in vacuo.
(S)-4-Phenyloxazolidin-2-one (11a)
Compound 11a was synthesized according to the general procedure, starting from (S)-2-amino-2-phenylethan-1-ol. The product was purified by flash column chromatography (silica gel) to afford 11a (230 mg, 78%) as white needle crystals; Rf = 0.42 (hexane/EtOAc 1:1). Analytical data are in agreement with those reported in the literature.[6]
1H NMR (300 MHz, CDCl3): δ = 7.20 (m, 5 H, arom.), 3.87 (m, 1 H, CH), 3.56 (m, 1 Hb, CH2), 3.46 (m, 1 Ha, CH2), 2.84 (br s, 3 H).
(S)-4-Isopropyloxazolidin-2-one (11b)
Compound 11b was synthesized according to the general procedure, starting from (S)-2-amino-3-methylbutan-1-ol. The product was purified by flash column chromatography (silica gel) to afford 11b (794 mg, 63%) as white needle crystals; Rf = 0.38 (hexane/EtOAc 1:1). Analytical data are in agreement with those reported in the literature.[6]
1H NMR (300 MHz, CDCl3): δ = 7.00 (s, 1 H), 4.42–4.48 (t, J = 8.4 Hz, 1 H), 4.11 (dd, J = 8.4, 6.3 Hz, 1 H), 3.61 (q, J = 6.9 Hz, 1 H), 1.70–1.76 (m, 1 H), 0.97 (d, J = 6.9 Hz, 3 H), 0.90 (d, J = 6.9 Hz, 3 H).
(S)-4-tert-Butyloxazolidin-2-one (11c)
Compound 11c was synthesized according to the general procedure, starting from (S)-2-amino-3,3-dimethylbutan-1-ol. The product was purified by flash column chromatography (silica gel) to afford 11c (823 mg, 53%) as white needle crystals; Rf = 0.35 (hexane/EtOAc 1:1). Analytical data are in agreement with those reported in the literature.[6]
1H NMR (300 MHz, CDCl3): δ = 6.81 (br s, 1 H), 4.36 (t, J = 8.8 Hz, 1 H), 4.18 (dd, J = 8.8, 5.6 Hz, 1 H), 3.60 (m, 1 H), 0.91 (s, 9 H).
Chloroacetyl Derivatives 12a–c; General Procedure
Freshly washed NaH (1.1 equiv.) was loaded in a heat gun-dried Schlenk tube under a N2 atmosphere, then freshly distilled THF was added (0.24 M), and the mixture was cooled to 0 °C. After that, the oxazolidinone 11 (1 equiv.) was added portionwise, and the reaction was warmed to r.t. The mixture was stirred for 5 h, then cooled at 0 °C. Chloroacetyl chloride (1.7 equiv.) was then added dropwise, and the mixture was stirred for 15 h at r.t. After this time, the mixture was filtered on a Celite® pad, and the solvent was evaporated under reduced pressure. The crude residue was then diluted in DCM, and the mixture was washed with 10% NaHCO3 solution, dried (MgSO4), filtered, and dried in vacuum. The target product was isolated after chromatographic purification (hexane/EtOAc 7:3).
(S)-3-(2-Chloroacetyl)-4-phenyloxazolidin-2-one (12a)
Synthesized according to the general procedure starting from (S)-4-phenyloxazolidin-2-one (11a). The product was purified by flash column chromatography (silica gel) to afford 12a (1.243 g, 92%) as a white solid; Rf = 0.28 (hexane/EtOAc 7:3). Analytical data are in agreement with those reported in literature.[6]
1H NMR (300 MHz, CDCl3): δ = 7.48–7.30 (m, 5 H), 5.46 (dd, J = 8.7, 3.8 Hz, 1 H), 4.84–4.69 (m, 3 H), 4.39 (dt, J = 6.1, 3.0 Hz, 1 H).
(S)-3-(2-Chloroacetyl)-4-isopropyloxazolidin-2-one (12b)
Synthesized according to the general procedure starting from (S)-4-isopropyloxazolidin-2-one (11b). The product was purified by flash column chromatography (silica gel) to afford 12b (1.384 g, 87%) as a white solid; Rf = 0.29 (hexane/EtOAc 7:3). Analytical data are in agreement with those reported in literature.[6]
1H NMR (300 MHz, CDCl3): δ = 4.81–4.64 (m, 2 H), 4.51–4.43 (m, 1 H), 4.37 (t, J = 8.6 Hz, 1 H), 4.29 (dd, J = 9.1, 3.2 Hz, 1 H), 2.58–2.27 (m, 1 H), 0.93 (dd, J = 13.2, 7.0 Hz, 6 H).
(S)-4-tert-Butyl-3-(2-chloroacetyl)oxazolidin-2-one (12c)
Synthesized according to the general procedure, starting from (S)-4-tert-butyloxazolidin-2-one (11c). The product was purified by flash column chromatography (silica gel) to afford 12c (300 mg, 76%) as a yellow oil; Rf = 0.26 (hexane/EtOAc 8:2). Analytical data are in agreement with those reported in the literature.[6]
1H NMR (300 MHz, CDCl3): δ = 4.73 (dd, J = 25, 15.46 Hz, 2 H), 4.45 (dd, J = 6.96, 2.2 Hz, 1 H), 4.37–4.29 (m, 2 H), 0.96 (s, 9 H).
Ylide Derivatives 13a–c; General Procedure
PPh3 (1.1 equiv.) was added to a solution of the chloroacetyl derivative 12 (1 equiv.) in degassed DCM (0.12 M), and the mixture was stirred at r.t. for 48 h under N2. The mixture was concentrated in vacuo and the residue was dissolved in hot water, 50 °C. After the addition of aq 4 M NaOH solution, the mixture was shaken for 5 min, immediately extracted with EtOAc, and the organic layer was washed with brine. The organic layers were combined, dried (anhyd Na2SO4), filtered, and concentrated in vacuo. The target product was isolated after chromatographic purification (typically hexane/EtOAc 7:3). Data are in agreement with literature.[6]
(S)-4-Phenyl-3-[2-(triphenylphosphanylidene)acetyl]oxazolidin-2-one (13a)
Synthesized according to the general procedure, starting from compound 12a. 13a was obtained as a foaming oil in 72% yield.
1H NMR (300 MHz, CDCl3): δ = 7.53–7.28 (m, 20 H), 5.59 (dd, J = 7.91, 2.53 Hz, 1 H), 4.62 (t, J = 7.34 Hz, 1 H), 4.15 (dd, J = 8.23, 3.47 Hz, 1 H).
31P NMR (122 MHz, CDCl3): δ = 18.18.
(S)-4-Isopropyl-3-[2-(triphenylphosphanylidene)acetyl]oxazolidin-2-one (13b)
Synthesized according to the general procedure, starting from compound 12b. 13b was obtained as a foaming oil in 81% yield.
1H NMR (300 MHz, CDCl3): δ = 7.74–7.40 (m, 15 H), 4.46 (td, J = 8.3, 4.3 Hz, 1 H), 4.29–4.03 (m, 3 H), 2.43–2.21 (m, 1 H), 0.92 (dd, J = 6.8, 3.7 Hz, 6 H).
31P NMR (122 MHz, CDCl3): δ = 18.02.
(S)-4-tert-Butyl-3-[2-(triphenylphosphanylidene)acetyl]oxazolidin-2-one (13c)
Synthesized according to the general procedure, starting from compound 12c. 13c was obtained as a foaming oil in 73% yield.
1H NMR (300 MHz, CDCl3): δ = 7.70–7.41 (m, 15 H), 4.48 (dd, J = 7.33, 2.30 Hz, 1 H), 4.22–4.10 (m, 3 H), 0.91 (s, 9 H).
31P NMR (122 MHz, CDCl3): δ = 17.93.
(2E,2′E)-4,4′-Oxybis(1-phenylbut-2-en-1-one) (7)
A solution of 2,5-dihydrofuran (3; 330 mg, 4.7 mmol, 1 equiv.) in DCM (16 mL) was placed in a 50-mL three-necked flask and cooled to –78 °C. Ozone (0.7 bar, 0.35 A) was passed through the mixture until a blue coloration persisted, at which point N2 was bubbled through the solution to remove excess dissolved ozone. The ozonide was then quenched with Me2S (700 μL, 9.53 mmol, 2.03 equiv.) and 2 (4.5 g, 11.7 mmol, 2.5 equiv) dissolved in DCM (10 mL), which was added dropwise at –78 °C under vigorous stirring. The resulting solution was warmed at r.t. and allowed to stir for 24 h. Concentration in vacuo and purification by chromatography (silica gel, hexane/EtOAc 9:1, 8:2, 7:3; Rf = 0.33 hexane/EtOAc 7:3) gave the final product 7 (695 mg, 2.3 mmol, 48%) as a pale-yellow oil. Analytical data are in agreement with those reported in the literature.[5]
1H NMR (300 MHz, CDCl3): δ = 7.99 (dt, J = 8.9, 2.9 Hz, 4 H), 7.24 (dt, J = 15.1, 1.8 Hz, 2 H), 7.06 (dt, J = 15.4, 4.1 Hz, 4 H), 6.95 (dt, J = 8.9, 2.9 Hz, 4 H), 4.36 (dd, J = 4.1, 2.0 Hz, 4 H).
13C NMR (75 MHz, CDCl3): δ = 188.3, 163.5, 142.7, 130.9, 130.5, 124.9, 113.8, 70.0, 55.5.
tert-Butyl Bis[(E)-4-oxo-4-phenylbut-2-en-1-yl]carbamate (8)
A solution of N-Boc-2,5-dihydropyrrole (4; 400 mg, 2.364 mmol, 1 equiv.) in DCM (8 mL) was placed in a 50-mL three-necked flask and cooled to –78 °C. Ozone (0.7 bar, 0.35 A) was passed through the mixture until a blue coloration persisted, at which point N2 was bubbled through the solution to remove excess ozone. The ozonide was then quenched with Me2S (352.4 μL, 4.8 mmol, 2.03 equiv.) and 2 (2.25 g, 5.910 mmol, 2.5 equiv.) dissolved in DCM (5 mL), which was added dropwise at –78 °C under vigorous stirring. The resulting solution was warmed at r.t. and allowed to stir for 24 h. Concentration in vacuo and purification by chromatography (silica gel, n-pentane/EtOAc 9:1, 8:2; Rf = 0.30 hexane/EtOAc 7:3) gave the final product 8 (275 mg, 0.678 mmol, 29%) as a yellow oil. For analytical data see the Supporting Information.
1H NMR (300 MHz, CDCl3): δ = 7.93 (d, J = 7.20 Hz, 4 H), 7.62–7.44 (dt, J = 31.46, 7.41 Hz, 6 H), 6.96 (s, 4 H), 4.25–4.07 (d, J = 21.53 Hz, 4 H), 1.51–1.49 (s, 9 H).
13C NMR (75 MHz, CDCl3): δ = 191.23, 155.23, 135.62, 133.09, 128.37, 126.73, 80.53, 53.44, 39.96, 29.67, 28.36.
Synthesis of Bisenones Derivatives 15a–c, 17; General Procedure
In a 25-mL round-bottomed flask equipped with a magnetic stirring bar, 14 or 16 (1 equiv.) was dissolved in dry DCM (0.5 mL) under N2 atmosphere. Anhyd MgSO4 (439 mg, 3.65 mmol, 9 equiv.) was then added. A solution of ylide 13 (1.3 equiv.) in dry DCM (1.2 mL) was added to the mixture, and the mixture was stirred for 48 h at r.t. The crude was then filtered through a pad of Celite® to remove the MgSO4 and concentrated in vacuo. Purification via flash chromatography yielded the final product. For analytical data see the Supporting Information.
tert-Butyl {(E)-4-Oxo-4-[(S)-2-oxo-4-phenyloxazolidin-3-yl]but-2-en-1-yl}[(E)-4-oxo-4-phenylbut-2-en-1-yl]carbamate (15a)
Synthesized starting from 14 and 13a using the general procedure and with purification using hexane/EtOAc 8:2 to 7:3; Rf = 0.20 (hexane/EtOAc 7:3). 15a was obtained as a pale yellow oil in 31% yield.
tert-Butyl {(E)-4-[(S)-4-Isopropyl-2-oxooxazolidin-3-yl]-4-oxobut-2-en-1-yl}[(E)-4-oxo-4-phenylbut-2-en-1-yl]carbamate (15b)
Synthesized starting from 14 and 13b using the general procedure and with purification using hexane/EtOAc 8:2, 7:3; Rf = 0.20 (hexane/EtOAc 7:3). 15b was obtained as a pale yellow oil in 23% yield.
tert-Butyl {(E)-4-[(S)-4-tert-Butyl-2-oxooxazolidin-3-yl]-4-oxobut-2-en-1-yl}[(E)-4-oxo-4-phenylbut-2-en-1-yl]carbamate (15c)
Synthesized starting from 14 and 13c, using the general procedure and with purification using hexane/EtOAc 8:2 to 7:3; Rf = 0.30 (hexane/EtOAc 7:3). 15c was obtained as a pale yellow oil in 48% yield.
(S)-4-tert-Butyl-3-((E)-4-{[(E)-4-oxo-4-phenylbut-2-en-1-yl]oxy}but-2-enoyl)oxazolidin-2-one (17)
Synthesized starting from 16 and 13c using general procedure and with purification using hexane/EtOAc 7:3 to 6:4; Rf = 0.41 (hexane/EtOAc 1:1). 17 was obtained as a colorless oil in 52% yield.
Photocatalytic Cycloaddition; General Procedure
In a 3-mL vial, Hantzsch ester (46.4 mg, 0.150 mmol, 1.1 equiv.), Schreiner’s thiourea (13.5 mg, 0.027 mmol, 0.2 equiv.), and Eosin Y (2 mg, 0.003 mmol, 0.025 equiv.), were added and three vacuum/nitrogen cycles were performed. Subsequently, distilled DIPEA (18 mg, 0.136 mmol, 1 equiv) and the bisenone 7, 8, 15a–c, or 17 (1 equiv.) dissolved in DCM (0.136 M), previously degassed for 30 min, were added to the reaction vial. The mixture was then irradiated for 5 h at a distance of 2 cm. After completion of the reaction (TLC), the solvent was removed and the product was purified, by chromatographic column (silica gel).
2,2′-(Tetrahydrofuran-3,4-diyl)bis(1-phenylethan-1-one) (9)
Synthesized starting from 7 using the general procedure with purification using hexane/EtOAc 9:1, 8:2; Rf = 0.30 (hexane/EtOAc 7:3). 9 was obtained as a colorless oil in 89% yield. Analytical data are in agreement with those reported in the literature.[5]
1H NMR (300 MHz, CDCl3): δ = 7.97–7.89 (m, 4 H), 7.54 (d, J = 7.4 Hz, 2 H), 7.44 (dd, J = 10.8, 4.4 Hz, 4 H), 4.15 (dd, J = 8.9, 6.7 Hz, 2 H), 3.48 (dd, J = 9.0, 5.9 Hz, 2 H), 3.37 (dd, J = 17.7, 5.1 Hz, 2 H), 3.09 (dd, J = 17.8, 8.2 Hz, 2 H), 2.60–2.50 (m, 2 H).
13C NMR (75 MHz, CDCl3): δ = 199.1, 136.8, 133.3, 128.7, 128.0, 73.4, 42.6, 40.6.
MS (APCI+): m/z [M + H]+ calcd for C20H20O3: 309.1; found: 308.9.
tert-Butyl 3,4-Bis(2-oxo-2-phenylethyl)pyrrolidine-1-carboxylate (10)
Synthesized starting from 8 using the general procedure with purification using hexane/EtOAc 9:1 to 8:2; Rf = 0.35 (hexane/EtOAc 7:3). 10 was obtained as a colorless oil in 51% yield.
1H NMR (300 MHz, CDCl3): δ = 7.98–7.83 (d, J = 6.38 Hz, 4 H), 7.60–7.38 (m, 6 H), 3.87–3.73 (dd, J = 10.32, 4.92 Hz, 2 H), 3.34–2.92 (m, 6 H), 2.66–2.47 (m, 2 H), 1.44 (s, 9 H).
13C NMR (75 MHz, CDCl3): δ = 198.54, 154.35, 136.72, 133.26, 128.65, 127.99, 79.24, 77.56, 77.14, 76.71, 68.51, 51.34, 50.91, 49.22, 42.99, 41.41, 39.59, 38.70, 28.51, 28.07, 25.01.
For analytical data see the Supporting Information.
tert-Butyl 3-{2-[(S)-4-tert-Butyl-2-oxooxazolidin-3-yl]-2-oxoethyl}-4-(2-oxo-2-phenylethyl)pyrrolidine-1-carboxylate (18c)
Synthesized starting from 15c using the general procedure with purification using hexane/EtOAc gradient from 8:2 to 7:3; Rf = 0.20 (hexane/EtOAc 7:3). 18c was obtained as a pale yellow oil in 42% yield. This material was used immediately in the next step.
1H NMR (300 MHz, CDCl3): δ = 7.91 (d, J = 7.7 Hz, 1 H), 7.51 (dt, J = 29.4, 7.4 Hz, 1 H), 7.33 (d, J = 14.7 Hz, 2 H), 6.90 (d, J = 4.3 Hz, 1 H), 4.49 (d, J = 7.4 Hz, 1 H), 4.39–4.20 (m, 2 H), 4.11 (q, J = 6.4 Hz, 1 H), 1.46 (d, J = 5.6 Hz, 8 H), 0.93 (d, J = 5.2 Hz, 9 H).
13C NMR (75 MHz, CDCl3): δ = 198.5, 171.6, 154.8, 154.3, 136.6, 133.3, 128.7, 128.2, 128.0, 126.3, 79.3, 77.5, 76.6, 65.6, 61.1, 53.4, 51.1, 41.3, 39.2, 38.5, 35.8, 29.7, 28.5, 28.3, 25.6, 23.8.
Removal of the Chiral Auxiliary; General Procedure
In a 3-mL vial, equipped with a magnetic stirring bar, under N2 atmosphere, the cyclized product (1 equiv.) was dissolved in dry MeOH (0.15 M). The mixture was then cooled to –20 °C and freshly prepared NaOMe solution (1.2 equiv.) was added under stirring. The mixture was stirred at –20 °C for 18 h. After completion of the reaction, the crude was allowed to warm to r.t. and then was quenched with 1 M HCl. The aqueous phase was extracted with DCM and the organic phase was washed with brine, dried (Na2SO4), and concentrated in vacuo. Purification by flash chromatography (silica gel, hexane/EtOAc 8:2 to 7:3) gave the desired product.
tert-Butyl 3-(2-Methoxy-2-oxoethyl)-4-(2-oxo-2-phenylethyl)pyrrolidine-1-carboxylate (20)
Synthesized starting from 18a–c using the general procedure. 20 was obtained as a colorless oil (Rf = 0.30 hexane/EtOAc 7:3). For analytical data and HPLC traces see the Supporting Information.
HPLC conditions: CHIRALCEL OJ-H column, hexane/i-PrOH 9:1, 0.8 mL/min, P = 36 bar.
Methyl 2-(4-(2-Oxo-2-phenylethyl)tetrahydrofuran-3-yl)acetate (21)
Synthesized starting from 19 using the general procedure. 21 was obtained as a colorless oil in 64% yield; Rf = 0.35 (hexane/EtOAc 7:3). For analytical data and HPLC traces see the Supporting Information.
HPLC conditions: CHIRALCEL OJ-H column, hexane/i-PrOH 9:1, 0.8 mL/min, P = 36 bar
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
The authors thank Ms. G. Bonfanti for contributions in preliminary experimental activity.
Supporting Information
- Supporting information for this article is available online at https://doi.org/10.1055/a-2766-4857.
- Supporting Information (PDF) (opens in new window)
-
References
- 1a Skubi KL, Blum TR, Yoon TP. Chem. Rev. 2016; 116: 10035
- 1b Candish L, Collins KD, Cook GC, Douglas JJ, Gomez-Suarez A, Jolit A, Keess S. Chem. Rev. 2022; 122: 2907
- 1c Romero NA, Nicewicz DA. Chem. Rev. 2016; 116: 10075
- 1d Prier CK, Rankic DA, MacMillan DW. Chem. Rev. 2013; 113: 5322
- 2 Wang L.-C, Jang H.-Y, Roh Y, Lynch V, Schultz AJ, Wang X, Krische MJ. J. Am. Chem. Soc. 2002; 124: 9448
- 3 Yang J, Felton GA. N, Bauld NL, Krische MJ. J. Am. Chem. Soc. 2004; 126: 1634
- 4 Du J, Espelt LR, Guzei IA, Yoon TP. Chem. Sci. 2011; 2: 2115
- 5 Neumann M, Zeitler K. Chem. Eur. J. 2013; 19: 6950
- 6a Medici F, Resta S, Presenti P, Caruso L, Puglisi A, Raimondi L, Rossi S, Benaglia M. Eur. J. Org. Chem. 2021; 2021: 4521
- 6b Resta S, Benettin T, Puglisi A, Raimondi L, Rossi S. Tetrahedron Lett. 2024; 138: 154976
- 7 Benettin T, Resta S, Forni A, Raimondi L, Puglisi A, Rossi S. Molecules 2025; 30: 2090
- 8 Benettin T, Resta S, Puglisi A, Rossi S. Eur. J. Org. Chem. 2024; 27: e202400816
- 9 Mahmoud N, Awassa J, Toufaily J, Lebeau B, Daou TJ, Cormier M, Goddard JP. Molecules 2023; 28: 549
- 10 Herbrik F, Camarero González P, Krstic M, Puglisi A, Benaglia M, Sanz MA, Rossi S. Appl. Sci. 2020; 10: 5596
- 11 Zhang XN, Shi M. Eur. J. Org. Chem. 2012; 2012: 6271
Corresponding Author
Publication History
Received: 04 November 2025
Accepted after revision: 05 December 2025
Accepted Manuscript online:
05 December 2025
Article published online:
26 January 2026
© 2025. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References
- 1a Skubi KL, Blum TR, Yoon TP. Chem. Rev. 2016; 116: 10035
- 1b Candish L, Collins KD, Cook GC, Douglas JJ, Gomez-Suarez A, Jolit A, Keess S. Chem. Rev. 2022; 122: 2907
- 1c Romero NA, Nicewicz DA. Chem. Rev. 2016; 116: 10075
- 1d Prier CK, Rankic DA, MacMillan DW. Chem. Rev. 2013; 113: 5322
- 2 Wang L.-C, Jang H.-Y, Roh Y, Lynch V, Schultz AJ, Wang X, Krische MJ. J. Am. Chem. Soc. 2002; 124: 9448
- 3 Yang J, Felton GA. N, Bauld NL, Krische MJ. J. Am. Chem. Soc. 2004; 126: 1634
- 4 Du J, Espelt LR, Guzei IA, Yoon TP. Chem. Sci. 2011; 2: 2115
- 5 Neumann M, Zeitler K. Chem. Eur. J. 2013; 19: 6950
- 6a Medici F, Resta S, Presenti P, Caruso L, Puglisi A, Raimondi L, Rossi S, Benaglia M. Eur. J. Org. Chem. 2021; 2021: 4521
- 6b Resta S, Benettin T, Puglisi A, Raimondi L, Rossi S. Tetrahedron Lett. 2024; 138: 154976
- 7 Benettin T, Resta S, Forni A, Raimondi L, Puglisi A, Rossi S. Molecules 2025; 30: 2090
- 8 Benettin T, Resta S, Puglisi A, Rossi S. Eur. J. Org. Chem. 2024; 27: e202400816
- 9 Mahmoud N, Awassa J, Toufaily J, Lebeau B, Daou TJ, Cormier M, Goddard JP. Molecules 2023; 28: 549
- 10 Herbrik F, Camarero González P, Krstic M, Puglisi A, Benaglia M, Sanz MA, Rossi S. Appl. Sci. 2020; 10: 5596
- 11 Zhang XN, Shi M. Eur. J. Org. Chem. 2012; 2012: 6271
