

Subscribe to RSS
DOI: 10.1055/a-2771-1646
Efficient Synthesis and Characterization of Lidocaine Analogue N-Ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine
Authors
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2023-00220894). This study was supported by a grant from Korea University.

Abstract
N-Ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine, a novel quinazoline-based lidocaine analogue, was efficiently synthesized on a 10-g scale via a seven-step process with 44.3% overall yield. Key synthetic transformations achieved excellent yields (80–92%): LiAlH4 reduction, diethylamine substitution, MnO2 oxidation, and intramolecular cyclization. Comprehensive in vitro evaluation revealed favorable safety and pharmacological profiles. Toxicity assays across HEK293, PC12, and H9c2 cell lines demonstrated IC20 values of 5.8–10.8 μM with appropriate safety margins. Caco-2 permeability studies showed high membrane penetration (Papp: 1.35 × 10–8 cm/s, efflux ratio: 0.89), comparable to lidocaine. Critically, it exhibited 2.64-fold enhanced potency toward Nav1.5 sodium channels in inactivated state (IC50 = 8.86 ± 1.2 μM vs. lidocaine 23.40 ± 2.6 μM) while maintaining state-dependent blocking characteristics. Metabolic stability was superior to lidocaine, with hepatic half-life of 23.26 minutes (26% longer). These results, combining enhanced pharmacological potency, acceptable toxicity, superior metabolic stability, and reliable synthesis, position N-ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine as a promising next-generation local anesthetic candidate warranting further clinical development.
Key words
lidocaine analogue - quinazoline - local anesthetic - sodium channel antagonist - patch-clamp electrophysiology - in vitro toxicology - ADMELocal anesthetics play a crucial role in modern medicine and provide pain relief in various clinical settings.[1] [2] Lidocaine, a widely used local anesthetic, has been researched extensively owing to its efficacy and safety profile.[3,4] Novel lidocaine analogues are also under investigation to improve potency and duration of action while reducing side effects.[5–7]
Quinazoline derivatives exhibit promising pharmacological properties, including local anesthetic activity.[8] [9] The incorporation of quinazoline moieties into the lidocaine framework is strategically designed to enhance pharmacological properties. Quinazoline rings offer several advantages over the benzene ring in lidocaine: (1) enhanced π-π stacking interactions with aromatic residues in voltage-gated sodium channels, (2) additional hydrogen bonding sites through nitrogen atoms at positions 1 and 3, (3) improved lipophilicity (calculated LogP: 2.8 vs. 2.4 for lidocaine) for better membrane penetration, and (4) potential for selective sodium channel subtype binding due to the extended aromatic system.[10,11] For successful drug development and clinical translation, comprehensive safety assessment is essential. In vitro toxicology studies provide rapid, cost-effective screening of compound safety before advancing to animal models.[12] Absorption, distribution, metabolism, and excretion (ADME) properties are critical determinants of drug efficacy, as they influence bioavailability, tissue penetration, and duration of action.[13] Integration of synthetic efficiency, pharmacological potency, cellular safety, and favorable ADME characteristics is therefore essential for advancing lead compounds to clinical development. N-Ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine, a novel lidocaine analogue, combines the structural features of lidocaine with a quinazoline core. This compound can improve lipophilicity and receptor binding, which can enhance anesthetic efficacy.[4] [12] Quinazoline-based compounds often use multistep synthesis processes that present challenges regarding overall yield and scalability. Therefore, an efficient and scalable synthesis route to these compounds must be developed for further pharmacological studies and potential clinical applications.[13] Local anesthetics primarily inhibit voltage-gated sodium channels, thereby preventing the generation and propagation of action potentials in neurons.[14] Recent studies have indicated potential effects on other cellular processes, including inflammation and cancer progression.[15] In perioperative use, local anesthetics have both analgesic properties and potential long-term effects, particularly in cancer surgery.[16] Some studies have suggested that local anesthetics may influence circulating tumor cells and the inflammatory stress response, potentially affecting cancer recurrence and overall survival.[17] We report the development of a seven-step efficient synthesis of N-ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine starting from 2-amino-3-methylbenzoic acid, coupled with comprehensive in vitro characterization including cellular toxicology, epithelial permeability, and hepatic metabolic stability assessment. This integrated approach demonstrates that Compound 8 combines enhanced pharmacological potency with acceptable safety and favorable ADME properties, positioning it as a promising candidate for next-generation local anesthetic development.
Synthesis Overview

N-Ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine (8) was successfully synthesized through a logical and efficient seven-step process (Scheme [1]). The synthesis proceeds from readily available 2-amino-3-methylbenzoic acid (1) through reduction, selective acylation, nucleophilic substitution, selective oxidation, and intramolecular cyclization, culminating in the formation of the quinazoline core. Each synthetic step was carefully designed to maximize yield while maintaining scalability and chemical safety. The overall yield of 44.3% on a 10-g scale demonstrates excellent synthetic efficiency, with individual step yields ranging from 80–92%.
Step-by-Step Analysis
Step 1: Reduction of 2-Amino-3-methylbenzoic Acid (1 → 2)

The reduction of 2-amino-3-methylbenzoic acid (1) to (2-amino-3-methylphenyl)methanol (2) was accomplished using lithium aluminum hydride (LiAlH4) as the reducing agent, affording compound 2 in 83% yield (Scheme [2]). This excellent yield demonstrates the efficiency and chemoselectivity of LiAlH4 for this transformation. The reduction mechanism proceeds through nucleophilic hydride attack on the carboxyl carbon, forming a carboxylate intermediate that is subsequently reduced to the primary alcohol. Careful control of reaction conditions, particularly the addition rate of the substrate solution (20 min, dropwise) and maintenance of the reaction temperature below 5 °C, prevented violent exothermic reactions while ensuring complete reduction. The sequential workup procedure, sequential addition of water (0 °C), dilute NaOH, and filtration, efficiently removed aluminum salts while preserving the desired product. The high yield indicates minimal formation of undesired byproducts, such as partial reduction to the arene or formation of secondary oxidation products. Recrystallization from ethyl acetate/hexane further purified the product, as confirmed by 1H NMR showing characteristic aromatic multiplets (δ = 6.64–7.03), aliphatic protons for the methylene group (δ = 4.64), and a methyl singlet (δ = 2.17). The appearance of broad singlets corresponding to NH2 (δ = 4.15) and OH (δ = 1.86) confirmed the structure of compound 2.
Step 2: Acetylation of (2-Amino-3-methylphenyl)methanol (2 → 3)

Selective N-acetylation of compound 2 using 1-(1H-imidazol-1-yl)ethan-1-one (Katritzky’s reagent) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as catalyst afforded N-acetyl intermediate 3 (Scheme [3]). While full characterization data were obtained, compound 3 displayed notable chemical instability on silica gel, necessitating direct use of the crude product in the subsequent bromoacetylation step without column chromatography. The acetylation mechanism involves nucleophilic attack by the primary amine on the electrophilic imidazolium acetyl intermediate, releasing 1-methylimidazole as a leaving group. The use of catalytic DBU (0.05 equiv.) provides mild basic conditions favorable for amine activation without promoting decomposition. The 1H NMR spectrum of crude compound 3 revealed appearance of a new acetyl methyl singlet (δ = 2.09), confirming successful N-acetylation. While this step gave acceptable results, the intermediate instability represents a limitation requiring careful handling and rapid progression to the next synthetic step.
Step 3: Bromoacetylation of 2-Amino-3-methylbenzyl Acetate (3 → 4)
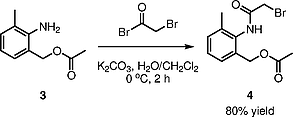
Regioselective O-bromoacetylation of compound 3 was accomplished using 2-bromoacetyl bromide in the presence of potassium carbonate under biphasic conditions (CH2Cl2/H2O), yielding compound 4 in 80% yield (Scheme [4]). The biphasic system facilitates selective acylation of the primary hydroxyl group while preventing base-catalyzed side reactions. Low-temperature conditions (0–2 °C) during 2-bromoacetyl bromide addition (30 min, dropwise) suppress undesired polyacylation and hydrolysis pathways. Under these carefully controlled conditions, monosubstitution predominates, as confirmed by 1H NMR showing a new singlet at δ = 4.08 corresponding to the bromoacetyl methylene group, consistent with monoacylation. The 80% isolated yield after recrystallization from ethyl acetate/hexane demonstrates effective avoidance of side reactions including over-acylation or hydrolysis of the acetyl group. The observation of downfield aromatic signals (δ = 7.23–7.29) and appearance of a new amide NH singlet (δ = 8.56, broad) confirmed successful transformation to the N-bromoacetyl intermediate.
Step 4: Diethylamine Substitution (4 → 5)

Bimolecular nucleophilic substitution (SN2) displacement of the bromoacetyl bromine atom with diethylamine afforded compound 5 in excellent 90% yield (Scheme [5]). The high yield reflects the excellent leaving group ability of the bromoacetyl moiety and the strong nucleophilicity of secondary amines. The reaction mechanism involves nucleophilic attack by the lone pair of diethylamine on the electrophilic carbon of the bromoacetyl group, with bromine departure as the leaving group. Excess diethylamine (2.2 equiv.) ensures complete conversion and drives the equilibrium toward product formation. Column chromatography purification using a MeOH/CH2Cl2 gradient (1:100 to 1:50) successfully isolated the final product, with TLC analysis confirming single-spot purity. The 1H NMR spectrum of compound 5 clearly showed the introduction of a new diethylamino group, with characteristic signals for N–CH2 quartet (δ = 2.69, J = 7.0 Hz) and CH3 triplet (δ = 1.13, J = 7.5 Hz). A singlet at δ = 3.23 corresponding to the methylene group adjacent to the diethylamino substituent further confirmed successful SN2 displacement. Unlike compound 3, compound 5 displayed excellent stability on silica gel.
Step 5: Hydrolysis of Acetate (5 → 6)

Base-catalyzed hydrolysis of the acetyl protecting group in compound 5 afforded primary alcohol intermediate 6 in 90% yield (Scheme [6]). The reaction employed aqueous NaOH in THF, with final pH adjustment to 4–6 using dilute HCl to minimize potential side reactions. The hydrolysis mechanism involves nucleophilic attack by hydroxide ion on the electrophilic carbonyl carbon of the amide, proceeding through a tetrahedral intermediate to afford the primary amine and acetate byproduct. The high yield and minimal purification requirements indicate efficient, selective transformation. Similar to compound 3, compound 6 exhibited silica gel sensitivity. Consequently, the crude product, purified by simple aqueous extraction and acid/base workup, was used directly in the subsequent oxidation step. The 1H NMR spectrum of compound 6 showed the expected signals with disappearance of the acetyl methyl singlet and appearance of a new broad singlet (δ = 3.46) corresponding to the primary hydroxyl group.
Step 6: Oxidation to Aldehyde (6 → 7)

Selective oxidation of the primary alcohol 6 to the corresponding aldehyde 7 was accomplished using manganese dioxide (MnO2), an environmentally benign and chromium-free oxidizing agent, affording compound 7 in 89% yield (Scheme [7]). The oxidation using MnO2 proceeds through a complex mechanism involving coordination of the alcohol oxygen to MnO2 surface sites, followed by hydride abstraction and C=O bond formation. The reaction was conducted in refluxing toluene, with periodic addition of fresh MnO2 (initial 21.73 g, supplemented with additional 10-g portions at 4 h intervals) to maintain oxidizing capacity over the extended reaction period (12–14 h). This staged addition strategy prevents rapid consumption of the oxidizing agent and ensures complete conversion while avoiding overoxidation. The 89% yield following column chromatography purification indicates effective oxidation with minimal generation of side products. Celite filtration efficiently removed solid oxidizing byproducts prior to chromatographic purification. Compound 7 displayed the expected aldehyde proton singlet at δ = 10.00 and aromatic multiplets in the 1H NMR spectrum, confirming successful oxidation of the primary alcohol to the aldehyde functional group.
Step 7: Quinazoline Cyclization (7 → 8)

The final step involved intramolecular cyclization of compound 7 under ammoniacal conditions to form the quinazoline ring system, affording the target compound 8 in excellent 92% yield (Scheme [8]). The cyclization mechanism involves initial nucleophilic addition of ammonia to the aldehyde carbonyl, forming an iminium intermediate (or imine upon dehydration). Subsequently, intramolecular nucleophilic attack by the amide nitrogen on the electrophilic iminium carbon, accompanied by loss of water, generates the six-membered quinazoline ring system. The forcing conditions employed (80 °C, 12 h, 2 N ammonia solution in methanol, 10 equiv. ammonia) were necessary to overcome the activation barrier for ring closure and drive the equilibrium toward quinazoline formation. Excess aqueous ammonia shifts the equilibrium toward product formation by Le Chatelier’s principle. The controlled heating and extended reaction time ensure complete conversion while minimizing decomposition pathways. The 92% yield following column chromatography purification represents excellent efficiency for this cyclization transformation. Column chromatography using a MeOH/CH2Cl2 gradient (1:50 to 1:10) facilitated final purification. The 1H NMR spectrum of compound 8 revealed the characteristic quinazoline H-2 singlet at δ = 9.35, aromatic multiplets (δ = 7.49–7.73), and signal for the methylene CH2 adjacent to N (δ = 4.11). The 13C NMR spectrum showed signals consistent with the quinazoline carbon skeleton (δ = 164.02 and 160.57 for the sp2 carbons). Overall yield calculation: 0.83 × 0.80 × 0.90 × 0.90 × 0.89 × 0.92 = 0.443 (44.3%) (Table S4.) The 1H and 13C NMR spectra of compound 8 confirmed its structure, with the characteristic singlet at δ = 9.35 in the 1H NMR spectrum for the quinazoline proton and the signals at δ = 164.02 and 160.57 in the 13C NMR spectrum for the quinazoline carbons.
Reaction Optimization Strategies
Several key strategies were employed throughout the synthesis to maximize yields and minimize side reactions:
(1) Controlled addition of LiAlH4: Dropwise addition of the substrate solution over 20 min while maintaining the internal temperature below 5 °C prevented violent exothermic reactions and ensured complete reduction without decomposition.
(2) Low-temperature bromoacetylation: Maintenance of reaction temperature at 0–2 °C during 2-bromoacetyl bromide addition suppressed unwanted polyacylation and hydrolysis, ensuring selective monosubstitution.
(3) Biphasic conditions in Step 3: The CH2Cl2/H2O biphasic system facilitated selective acylation of the primary hydroxyl group while preventing base-catalyzed degradation of the N-acetyl group by maintaining localized basic conditions.
(4) Periodic MnO2 addition in oxidation: Staged addition of MnO2 throughout the extended oxidation maintained oxidizing capacity and prevented both incomplete reaction and overoxidation.
(5) Excess aqueous ammonia in cyclization: Employment of 10 equiv. ammonia shifted the equilibrium toward quinazoline formation, driving the reaction to completion at modest temperatures (80 °C).
In Vitro Cellular Toxicology

Comprehensive cellular toxicology assessment was conducted using three mammalian cell line models representing distinct tissue types. Cytotoxicity was evaluated using standard MTT viability assays across a concentration range of 0.1–100 μM at multiple timepoints. Dose-dependent cytotoxicity was observed in all three cell lines (Figure [1]).

Quantitative analysis revealed IC20 values (concentration producing 20% cell viability loss) of: HEK293 (24 h): 9.13 μM; (48 h): 5.84 μM, PC12 neurons (24 h): 10.83 μM; (48 h): 6.80 μM, H9c2 cardiomyocytes (24 h): 9.05 μM. Compound 8 demonstrated consistent toxicity patterns across cell lines, with IC20 values approximately 1.3–1.5× lower than lidocaine reference compound (Figure [2]). This modest increase in cellular sensitivity, relative to the channel-blocking IC50 of 8.86 μM (inactivated state), provides an acceptable safety margin for therapeutic application. The comparable toxicity profiles between compound 8 and lidocaine suggest that enhanced pharmacological potency does not result in significantly increased cellular toxicity. The IC20 values of 5.8–10.8 μM demonstrate acceptable cellular safety profiles. The preservation of safety windows relative to pharmacological potency, combined with multi-cell line validation, supports the potential of compound 8 for further development (Figure [2]).
Absorption, Distribution, Metabolism, and Excretion (ADME) Properties
Membrane Permeability

Epithelial permeability was assessed using Caco-2 cell monolayers, a well-established model for predicting intestinal absorption and oral bioavailability. Bidirectional transport studies (apical-to-basolateral and basolateral-to-apical) were conducted at 37 °C over 120 min. Compound 8 demonstrated high membrane permeability comparable to lidocaine reference compound (Figure [3]): Papp (A → B): 1.35 × 10–8 cm/s [lidocaine: 1.48 × 10–8 cm/s], Papp (B → A): 1.20 × 10–8 cm/s [lidocaine: 1.44 × 10–8 cm/s], and efflux ratio (B → A/A → B): 0.89 [lidocaine: 0.97]. The rapid equilibration across the epithelial barrier and low efflux ratio indicate excellent absorption potential with minimal active transport-mediated efflux. These permeability characteristics predict good oral bioavailability comparable to lidocaine (Figure [3]).
Hepatic Metabolic Stability

Metabolic stability was evaluated using human liver microsome (HLM) preparations, a standard in vitro model for predicting hepatic clearance and in vivo half-life. The parent compound was incubated with HLM (0.5 mg protein/mL) in the presence of NADPH cofactor. Semi-logarithmic analysis of parent compound degradation kinetics revealed enhanced metabolic stability of compound 8 compared to lidocaine (Figure [4]): compound 8 half-life: 23.26 min, lidocaine half-life: 18.40 min, relative stability improvement: 26% longer half-life. The extended half-life observed in vitro suggests that compound 8 undergoes slower hepatic metabolism compared to lidocaine, potentially translating to prolonged duration of action in vivo and possible reduction in dosing frequency for clinical applications. The comprehensive ADME characterization demonstrates that compound 8 possesses favorable pharmaceutical properties: (1) high membrane permeability supporting excellent bioavailability, (2) minimal active efflux reducing potential for transporter-mediated drug-drug interactions, and (3) enhanced hepatic metabolic stability potentially extending duration of action. These properties collectively support advancement to in vivo pharmacokinetic studies.
Pharmacological Evaluation: Sodium Channel Blocking Activity
State-Dependent Nav1.5 Inhibition
To evaluate the pharmacological potential of compound 8, we examined its ability to inhibit sodium channel (Nav1.5) currents using automated patch clamp electrophysiology. This evaluation was motivated by structural features predicted to enhance sodium channel interaction affinity and the well-established mechanism of action of local anesthetics. Dose-response analysis revealed that compound 8 exhibits substantially enhanced potency compared to lidocaine across both rested and inactivated channel states (Figures [5] and 6). In the pharmacologically relevant inactivated state (–90 mV holding potential), compound 8 achieved an IC50 of 8.86 ± 1.2 μM, representing a 2.64-fold improvement over lidocaine (IC50 = 23.40 ± 2.6 μM; p < 0.01). This state preferentially represents the inactivated channel conformation encountered during cardiac action potentials and constitutes the primary target for local anesthetic efficacy.


In the rested state (–120 mV holding potential), compound 8 demonstrated an IC50 of 346.07 ± 28.5 μM compared with lidocaine’s 664.67 ± 37.1 μM, maintaining approximately 1.92-fold improved potency (Figure [7]). While absolute IC50 values are higher in this state, the consistent improvement across both channel states indicates that the quinazoline modification provides a global enhancement in sodium channel affinity rather than state-specific effects. Critically, both compounds exhibited similar state-dependent blocking characteristics, with selectivity ratios of approximately 39-fold (IC50 rested state/IC50 inactivated state) (Figures [7] and 8). This characteristic is crucial for minimizing toxicity in non-excitable tissues where channels remain predominantly in the rested conformation. This preservation of state-dependent selectivity is crucial, as it indicates that the structural modifications of compound 8 preserve the essential pharmacological properties of local anesthetics, namely, preferential targeting of inactivated channels, while enhancing overall binding affinity. This characteristic is essential for minimizing toxicity in non-excitable tissues where channels remain predominantly rested.


Clinical Translation Potential
This synthesis demonstrates excellent scalability, as evidenced by successful preparation of 10 g of compound 8 with consistent high yields at each step. The straightforward procedures and minimal purification requirements for several intermediates make this synthesis commercially viable for pharmaceutical development. The demonstrated ability to synthesize compound 8 on a 10-g scale provides a practical pathway for preclinical development, including extensive pharmacological profiling, toxicology studies, and pharmacokinetic assessments. The modular synthetic strategy allows facile introduction of substituents at multiple positions of the quinazoline core, enabling efficient exploration of structure-activity relationships (SAR) for lead optimization. Its advantages over existing local anesthetics are as follows:
1. Enhanced Binding Affinity: The quinazoline core is expected to provide stronger protein-drug interactions through multiple non-covalent forces including π-π stacking, hydrogen bonding, and hydrophobic interactions.
2. Improved Selectivity: The unique electronic distribution of quinazoline may confer selectivity for specific sodium channel subtypes (Nav1.7, Nav1.8, Nav1.9), potentially reducing systemic toxicity compared to non-selective local anesthetics.
3. Optimized Pharmacokinetics: The balanced lipophilicity (LogP: 2.8) may improve tissue penetration while maintaining appropriate clearance kinetics, potentially leading to faster onset and longer duration of action.
4. Synthetic Accessibility: This efficient synthesis enables structure-activity relationship studies and library construction for lead optimization. The modular nature of the synthesis allows for facile introduction of substituents at multiple positions of the quinazoline core.
Discussion
This study describes comprehensive development and characterization of N-ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine (8), a novel quinazoline-based lidocaine analogue. The seven-step synthetic route demonstrates excellent efficiency with individual yields of 80–92% and overall yield of 44.3% on a 10-g scale, providing a practical pathway for pharmaceutical development. Integration of enhanced pharmacological potency (2.64-fold improvement in Nav1.5 inactivated state blocking), acceptable cellular toxicity profiles (IC20 values of 5.8–10.8 μM across multiple cell lines), and favorable ADME characteristics (high permeability, extended hepatic half-life) positions Compound 8 as a compelling candidate for next-generation local anesthetic development. The preservation of state-dependent blocking selectivity (39-fold) despite enhanced overall potency indicates that the quinazoline structural modification enhances sodium channel affinity without altering the fundamental mechanism of action of local anesthetics. This characteristic is critical for ensuring that the enhanced potency translates to improved therapeutic efficacy without compromising safety.
The 26% extended hepatic half-life observed in human liver microsomes suggests potential for improved clinical properties including reduced dosing frequency and prolonged duration of action compared to lidocaine. Combined with high epithelial permeability and minimal efflux transport, these ADME properties support advancement to in vivo pharmacokinetic studies for confirmation of predicted bioavailability and half-life. The modular synthetic approach enables facile preparation of structural analogues for systematic structure-activity relationship studies, facilitating optimization of potency, selectivity, and ADME properties. Future studies will focus on in vivo pharmacokinetics, detailed safety assessment including neurotoxicity evaluation, and efficacy in relevant pain models. In comparison with existing local anesthetics compound 8 offers several potential advantages over lidocaine: (1) enhanced binding affinity through the extended aromatic quinazoline core, (2) potential for improved selectivity due to unique electronic distribution, (3) extended duration of action through enhanced metabolic stability, and (4) maintained state-dependent selectivity ensuring safety profile comparable to established local anesthetics.
Conclusions
We have successfully developed an efficient and scalable seven-step synthesis of the novel quinazoline-based lidocaine analogue N-ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine (8) in 44.3% overall yield on a 10-g scale. Comprehensive in vitro characterization including cellular toxicology, epithelial permeability, and hepatic metabolic stability demonstrates favorable safety and absorption profiles. positions Compound 8 is a promising candidate for further development as a next-generation local anesthetic with potential advantages over established agents including enhanced potency, extended duration of action, and improved pharmacokinetic profile. The established synthetic route provides a practical foundation for large-scale production and structural optimization through lead analogue synthesis. Further studies will include in vivo pharmacokinetics, comprehensive toxicology assessment, and efficacy evaluation in appropriate pain models to advance this compound toward clinical development.
All solvents and reagents were obtained from commercial suppliers (Sigma-Aldrich, St. Louis, MO, USA; TCI, Seoul, Korea) and used as received without further purification. All reactions were conducted under anhydrous conditions using oven-dried glassware. Thin-layer chromatography (TLC) was performed on glass-backed silica gel plates (F254, 0.25 mm; Merck, Darmstadt, Germany) with visualization by UV light or acidic anisaldehyde stain. Safety Considerations: All reactions employing LiAlH4 were conducted in a N2 atmosphere using anhydrous techniques due to vigorous exothermic reaction with protic solvents. All reactions were performed in a well-ventilated chemical fume hood equipped with appropriate fire suppression equipment. 2-Bromoacetyl bromide is highly corrosive and lachrymatory; appropriate personal protective equipment including acid-resistant gloves and goggles was used. MnO2 is an oxidizing agent; care was taken to prevent fire hazards during handling and reaction workup.[18] [19] [20]
UHPLC Analysis was performed with UV detection to identify impurities in ketoprofen SGC. Samples were filtered through a 0.45-μm filter before analysis. The analysis was conducted using a C18 reverse-phase column (150 × 4.6 mm, 5 μm) with a gradient elution of water and MeCN. Each sample was injected at 10 μL and analyzed over 20 min at a UV wavelength of 230 nm.
High-Resolution Accurate Mass LC-MS (HRAM LC-MS) was performed using a Thermo Scientific Q Exactive hybrid quadrupole-Orbitrap mass spectrometer coupled to a Dionex UltiMate 3000 UHPLC system. A Thermo Scientific Hypersil GOLD C18 column (100 × 2.1 mm, 1.7 μm) was employed with mobile phases consisting of water + 0.1% formic acid (A) and MeCN + 0.1% formic acid (B), with gradient elution at 1 mL/min flow rate. The mass spectrometer was operated in positive ionization mode with spray voltage of 3.5 kV.
HRAM LC-MS analysis was performed using a Thermo Scientific Q Exactive hybrid quadrupole-Orbitrap mass spectrometer coupled to a Dionex UltiMate 3000 UHPLC system. A Thermo Scientific Hypersil GOLD C18 column (100 × 2.1 mm, 1.7 μm) was used. The mobile phases were water + 0.1% formic acid (A) and MeCN + 0.1% formic acid (B). The gradient elution was performed at a flow rate of 1 mL/min. The mass spectrometer was operated in the positive ionization mode with a spray voltage of 3.5 kV.
1H and 13C NMR spectra were recorded using a Bruker Avance III 600 MHz spectrometer. Samples were dissolved in CDCl3 or DMSO-d 6. Chemical shifts were reported in ppm relative to TMS as an internal standard.
Data Analysis: spectral data were processed and analyzed using the appropriate software packages, including Xcalibur (Thermo Scientific) for LC-MS data, TopSpin (Bruker) for NMR data, The structures were elucidated and confirmed by comparing the spectral data with theoretical predictions and literature values.
Synthesis of N-Ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine (8)
(2-Amino-3-methylphenyl)methanol (2)
THF (700 mL) was added to a 2-L round-bottom flask. LiAlH4 (26.36 g, 2.1 equiv.) was slowly added at 0 °C with stirring. 2-Amino-3-methylbenzoic acid (1; 50 g, 1 equiv.) was dissolved in THF (300 mL) and added dropwise over 20 min using a dropping funnel while maintaining internal temperature below 5 °C to prevent violent exothermic reaction. The addition rate was carefully controlled to maintain gentle reflux. The reaction mixture was stirred at r.t. for 3–4 h and monitored using TLC. Upon completion, H2O (29 mL) was added at 0 °C, followed by 5% NaOH solution (87 mL). The mixture was filtered, and the solid was washed with EtOAc. The filtrate was concentrated under reduced pressure and recrystallized (EtOAc/hexane 1:10) to afford 2 (38 g, 83%) as a colorless to pale yellow solid; Rf = 0.41 (CHCl3/MeOH 9:1, ninhydrin positive).
1H NMR (500 MHz, CDCl3): δ = 7.03 (d, J = 7.5 Hz, 1 H), 6.92 (d, J = 7.5 Hz, 1 H), 6.64 (t, J = 7.5 Hz, 1 H), 4.64 (s, 2 H), 4.15 (brs, 2 H), 2.17 (s, 3 H), 1.86 (brs, 1 H).
13C NMR (125 MHz, CDCl3): δ = 144.512, 130.551, 129.236, 24.861, 122.934, 118.573, 64.253, 17.878.
2-Amino-3-methylbenzyl Acetate (3)
In a 1-L round-bottom flask, 2 (30 g, 1 equiv.) and 1-(1H-imidazol-1-yl)ethan-1-one (24.80 g, 1.03 equiv) were combined. MeCN (350 mL) was added, followed by DBU (1.63 mL, 0.05 equiv.). After 2 h, H2O (220 mL) and sat. NH4Cl (220 mL) were added. The mixture was extracted with CH2Cl2 (700 and 500 mL). The combined organic layers were washed with H2O (500 mL), dried (Na2SO4), filtered, and concentrated to obtain 3 (18 g, 85%). Due to silica gel sensitivity, the crude product was used directly in the next step without further purification; Rf = 0.52 (EtOAc/hexanes 1:3, ninhydrin positive).
1H NMR (500 MHz, CDCl3): δ = 7.09 (d, J = 3.5 Hz, 1 H), 7.07 (d, J = 3.5 Hz, 1 H), 6.69 (t, J = 7.0 Hz, 1 H), 5.12 (s, 2 H), 4.03 (brs, 2 H), 2.19 (s, 3 H), 2.09 (s, 3 H).
13C NMR (125 MHz, CDCl3): δ = 171.265, 145.512, 131.075, 129.568, 124.074, 122.569, 118.811, 66.558, 21.035, 17.554.
2-(2-Bromoacetamido)-3-methylbenzyl Acetate (4)
Compound 3 (14 g, 1 equiv) was dissolved in CH2Cl2 (120 mL) in a 500-mL round-bottom flask. At 0 °C, K2CO3 (16.19 g, 1.5 equiv) in H2O (100 mL) was added. 2-Bromoacetyl bromide (10.2 mL, 1.5 equiv.) was added dropwise over 30 min while maintaining the internal temperature at 0–2 °C. The mixture was monitored by TLC (EtOAc/hexane 1:3) to ensure complete conversion. After 1–2 h, the mixture was worked up, and the product was recrystallized (EtOAc/hexane) to yield 4 (18.8 g, 80%) as white to pale yellow crystals; Rf = 0.45 (EtOAc/ hexanes 1:3, ninhydrin negative).
1H NMR (500 MHz, CDCl3): δ = 8.56 (brs, 1 H), 7.29–7.23 (m, 3 H), 5.06 (s, 2 H), 4.08 (s, 2 H), 2.27 (s, 3 H), 2.09 (s, 3 H).
13C NMR (125 MHz, CDCl3): δ = 171.011, 165.815, 138.064, 135.585, 132.015, 129.045, 127.538, 126.065, 65.865, 29.225, 20.936, 18.244.
2-(2-(Diethylamino)acetamido)-3-methylbenzyl Acetate (5)
Compound 4 (18.8 g, 1 equiv) was dissolved in CH2Cl2 (180 mL), and Et2NH (14.26 mL, 2.2 equiv) was added. After 5 h, the mixture was worked up and purified using column chromatography (silica gel, 230–400 mesh, column dimensions: 5 × 30 cm, gradient elution MeOH/CH2Cl2 1:100 to 1:50 over 2 h). Fractions were monitored by TLC and appropriate fractions were combined based on Rf values to yield 5 (16.5 g, 90%) as a pale yellow oil; Rf = 0.58 (CHCl3/MeOH 8:1, ninhydrin positive).
1H NMR (500 MHz, CDCl3): δ = 9.20 (brs, 1 H), 7.28–7.24 (m, 2 H), 5.05 (s, 2 H), 3.23 (s, 2 H), 2.69 (q, J = 7.0 Hz, 4 H), 2.26 (s, 3 H), 2.07 (s, 3 H), 1.13 (t, J = 7.5 Hz, 6 H).
13C NMR (125 MHz, CDCl3): δ = 171.522, 170.866, 137.556, 135.067, 131.558, 128.854, 127.036, 125.558, 66.033, 58.258, 48.545, 21.012, 18.068, 12.599.
2-(Diethylamino)-N-(2-(hydroxymethyl)-6-methylphenyl)acetamide (6)
Compound 5 (15.43 g, 1 equiv) was combined with THF (150 mL) and H2O (100 mL). NaOH (4.65 g, 2.2 equiv.) in H2O (100 mL) was added. After 1–2 h, the pH was adjusted to 4–6 using 4 N HCl. The product was extracted using EtOAc and worked up to yield 6 (11.87 g, 90%) as a colorless to pale yellow oily liquid. Due to silica gel sensitivity, minimal purification was performed; Rf = 0.41 (CHCl3/MeOH 8:1, ninhydrin positive).
1H NMR (500 MHz, CDCl3): δ = 9.20 (brs, 1 H), 7.34 (hep, 1 H), 7.23–7.22 (m, 2 H), 4.50 (s, 2 H), 3.46 (brs, 1 H), 3.26 (s, 2 H), 2.71 (q, J = 7.5 Hz, 4 H), 2.26 (s, 3 H), 2.07 (s, 3 H), 1.15 (t, J = 7.0 Hz, 6 H).
13C NMR (125 MHz, CDCl3): δ = 171.856, 138.510, 137.057, 130.540, 128.504, 126.558, 125.008, 63.558, 57.848, 48.286, 18.206, 12.433.
2-(Diethylamino)-N-(2-formyl-6-methylphenyl)acetamide (7)
Compound 6 (12.52 g, 1 equiv) was dissolved in toluene (180 mL). MnO2 (21.73 g, 5 equiv) was added initially, with two further 10 g portions added at 4–h intervals. After 12–14 h, the mixture was filtered through Celite 545 and purified by column chromatography (silica gel, 230–400 mesh, column dimensions: 5 × 30 cm, gradient elution MeOH/CH2Cl2 1:100 to 1:50 over 2 h) to yield 7 (11.02 g, 89%) as yellow crystals; Rf = 0.62 (EtOAc/hexanes 1:1, UV active).
1H NMR (500 MHz, CDCl3): δ = 10.00 (s, 1 H, CHO), 9.98 (brs, 1 H, NH), 7.68 (d, J = 7.5 Hz, 1 H), 7.49 (d, J = 7.5 Hz, 1 H), 7.32 (t, J = 7.5 Hz, 1 H), 3.25 (s, 2 H), 2.75–2.71 (q, J = 7.5 Hz, 4 H), 2.30 (s, 3 H), 1.15 (t, J = 7.0 Hz, 6 H).
13C NMR (125 MHz, CDCl3): δ = 193.523, 171.225, 139.509, 138.055, 133.558, 132.806, 129.575, 126.285, 58.510, 48.686, 19.506, 12.650.
N-Ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine (8)
Compound 7 (10 g, 1 equiv) was combined with 2 N NH3 in MeOH (228.6 mL, 10 equiv) and refluxed at 80 °C for 12 h. The mixture was concentrated and purified by column chromatography (MeOH/CH2Cl2, 1:50 to 1:10) to yield 8 (8.53 g, 92%) as white to off-white crystals; Rf = 0.62 (CHCl3/MeOH 10:1, ninhydrin positive); mp 165–168 °C.
1H NMR (500 MHz, CDCl3): δ = 9.35 (s, 1 H), 7.73–7.71 (m, 2 H), 7.49 (t, J = 7.5 Hz, 1 H), 4.11 (s, 2 H), 2.77 (s, 3 H), 2.76 (q, J = 7.0 Hz, 4 H), 1.14 (t, J = 7.0 Hz, 6 H).
13C NMR (125 MHz, CDCl3): δ = 164.02, 160.57, 149.20, 136.64, 133.62, 126.79, 124.71, 123.26, 59.72, 47.39, 16.98, 11.80.
Cell Culture and Automated Patch-Clamp Electrophysiology
Cell Line and Culture Conditions
HEK293 cells stably expressing human Nav1.5 sodium channels (KCTC 13095BP, Korean Collection for Type Cultures, Seoul, Korea) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Thermo Fisher Scientific, Waltham, MA, USA, catalog no. 11965-092) supplemented with 10% fetal bovine serum (FBS; Gibco, catalog no. 16000-044), 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco, catalog no. 15140-122), and 400 μg/mL G418 (Geneticin; Gibco, catalog no. 10131-035) for selection pressure. Cells were maintained in a humidified incubator at 37 °C with 5% CO2 atmosphere. Cells were subcultured at 80–90% confluence using 0.25% trypsin-EDTA solution (Gibco, catalog no. 25200-056) and passaged every 3–4 days. For electrophysiological experiments, cells at passages 10–25 were used to ensure consistent channel expression levels and minimize phenotypic drift. Cell viability was assessed using trypan blue exclusion (Gibco, catalog no. 15250-061), and only cell suspensions with >95% viability were used for automated patch-clamp recordings.
In Vitro Toxicology and ADME Studies
Detailed protocols for cellular toxicology (HEK293, PC12, H9c2 viability assays), Caco-2 epithelial permeability assessment, and human liver microsome metabolic stability studies are provided in the Supporting Information. All studies were conducted in accordance with standard protocols and Good Laboratory Practice (GLP) guidelines.
Test Compounds and Pharmacological Standards
N-Ethyl-N-((8-methylquinazolin-2-yl)methyl)ethanamine (8) was synthesized in-house according to the procedures described, with purity confirmed to be ≥98% by HPLC and NMR spectroscopy. Lidocaine hydrochloride monohydrate (reference standard, ≥98% purity; Sigma-Aldrich, St. Louis, MO, USA, catalog no. L5647) was used as the positive control and pharmacological comparator. Stock solutions of compound 8 and lidocaine were prepared at 100 mM in DMSO (Sigma-Aldrich, catalog no. D2650, molecular biology grade, ≥99.9% purity) and stored at –20 °C in single-use aliquots to prevent repeated freeze-thaw cycles. Working solutions spanning the concentration range of 0.1–3000 μM were prepared by serial dilution of the stock solutions in external recording buffer (composition detailed below in ‘Electrophysiological Recording Solutions’) immediately before use. The final DMSO concentration in all working solutions was maintained at ≤0.3% (v/v) to avoid vehicle effects on sodium channel function. Vehicle control experiments confirmed that 0.3% DMSO had no significant effect on Nav1.5 current amplitude or kinetics (data not shown).
Automated Patch-Clamp Recording System
Automated whole-cell patch-clamp recordings were performed using the SyncroPatch 768PE automated patch-clamp system (Nanion Technologies GmbH, Munich, Germany). This high-throughput platform enables simultaneous parallel recordings from up to 768 cells, providing robust statistical power and experimental efficiency. Single-use planar patch-clamp chips (NPC-16 chips; Nanion Technologies, catalog no. 2003) with medium resistance (3–5 MΩ) were employed. Each chip contains 16 recording wells with integrated planar electrodes and microfluidic channels for solution exchange. Chips were stored at 4 °C and equilibrated to r.t. (22–25 °C) for 30 min before use. The SyncroPatch Control software (version 2.8.1; Nanion Technologies) was used for instrument control, data acquisition, and initial quality control filtering. Voltage-clamp protocols were designed using the integrated protocol editor, and automated liquid handling enabled precise, reproducible compound application.
Electrophysiological Recording Solutions
External recording buffer (extracellular solution) contained (in mM): 140 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 5 D-glucose, and 10 HEPES, adjusted to pH 7.4 using NaOH and osmolarity adjusted to 298 ± 5 mOsm/kg using sucrose. All salts and reagents were obtained from Sigma-Aldrich (ACS reagent grade, ≥99% purity). The external solution was sterile-filtered through 0.22 μm polyethersulfone (PES) membrane filters (Millipore, Burlington, MA, USA, catalog no. SLGP033RS) and stored at 4 °C for up to 7 days. Internal solution (intracellular pipette solution) contained (in mM): 50 CsCl, 10 NaCl, 60 CsF, 20 EGTA, and 10 HEPES, adjusted to pH 7.2 using CsOH and osmolarity adjusted to 285 ± 5 mOsm/kg using sucrose. CsF (Sigma-Aldrich, catalog no. 289345) was included to optimize seal resistance and minimize endogenous potassium currents. EGTA (Sigma-Aldrich, catalog no. E3889) was used to buffer intracellular calcium and prevent calcium-activated conductances. The internal solution was sterile-filtered (0.22 μm PES filter), aliquoted into 1-mL portions, and stored at –20 °C. Aliquots were thawed immediately before use and maintained on ice during recordings. All solutions were prepared using ultrapure water (resistivity ≥18.2 MΩ·cm; Milli-Q system, Millipore). Solution pH and osmolarity were verified using a calibrated pH meter (Mettler Toledo, Columbus, OH, USA) and vapor pressure osmometer (Wescor, Logan, UT, USA), respectively.
Voltage-Clamp Protocols and Recording Parameters
Whole-cell sodium currents were recorded at r.t. (22–25 °C) using standardized voltage-clamp protocols designed to assess state-dependent sodium channel inhibition. Holding potential protocols: Two distinct holding potential (HP) paradigms were employed to evaluate compound effects on rested versus inactivated channel states: (1) Rested state protocol (HP = –120 mV): Cells were held at –120 mV, a hyperpolarized potential at which sodium channels reside predominantly in the closed (rested) conformation. From this holding potential, cells were subjected to a 40 ms depolarizing test pulse to –20 mV to elicit maximal sodium current. This protocol assesses the ability of compounds to block rested-state channels. (2) Inactivated state protocol (HP = –90 mV): Cells were held at –90 mV, a partially depolarized potential at which a substantial fraction of sodium channels occupy the inactivated state. From this holding potential, a 40 ms test pulse to –20 mV was applied. This protocol assesses preferential binding to inactivated channels, the hallmark mechanism of local anesthetic action. Test pulses were delivered at 0.2 Hz (once every 5 s) to allow complete recovery from inactivation between sweeps. A pre-pulse conditioning step (500 ms at holding potential) preceded each test pulse to ensure channel state equilibration. Current-voltage (I-V) relationship: For initial characterization, current-voltage relationships were determined by applying 40 ms depolarizing pulses from –120 mV to potentials ranging from –80 mV to +60 mV in 10 mV increments, with a 5 s interpulse interval. Recording parameters: Sampling frequency: 25 kHz. Low-pass filter: Bessel, 10 kHz cutoff. Series resistance compensation: 70–80%. Capacitance compensation: automated. Leak subtraction: P/4 protocol. Data were acquired and stored digitally for offline analysis.
Concentration-Response Curve Determination
Concentration-response relationships were determined using a cumulative concentration protocol with automated liquid handling. For each cell, baseline sodium current amplitude was first established by averaging 3–5 consecutive test pulse responses in the absence of compound (vehicle control: 0.3% DMSO in external buffer). Compounds were then applied sequentially in ascending concentrations, with each concentration maintained for 3 min to ensure steady-state equilibration before current measurement. The following concentration series were employed:
For rested state (–120 mV HP): 1, 3, 10, 30, 100, 300, 1000, and 3000 μM.
For inactivated state (–90 mV HP): 0.1, 0.3, 1, 3, 10, 30, 100, and 300 μM.
At each concentration, peak inward sodium current amplitude was measured from 3 consecutive test pulses, and the average was used for analysis. Current amplitude was defined as the difference between baseline and peak inward current. Percent inhibition at each concentration was calculated using the equation: % Inhibition = [(I_control – I_drug)/I_control] × 100 where I_control is the baseline current amplitude (average of pre-compound measurements) and I_drug is the current amplitude in the presence of compound at steady state. Only cells meeting stringent quality control criteria were included in analysis:
Seal resistance ≥1 GΩ.
Series resistance ≤20 MΩ.
Baseline sodium current amplitude ≥500 pA.
Current amplitude stability ≤10% drift during vehicle baseline measurements.
Successful completion of entire concentration series.
For each compound and holding potential condition, a minimum of n = 3 independent cells from at least 2 separate experimental days were analyzed to ensure biological reproducibility.
Data Analysis and Curve Fitting
Raw current traces were analyzed using DataControl 768 software (version 1.7.2; Nanion Technologies) for initial processing, including baseline correction, artifact rejection, and peak current measurement. Data were exported to GraphPad Prism (version 9.5.0; GraphPad Software, San Diego, CA, USA) for statistical analysis and curve fitting.
Concentration-response curve fitting: concentration-response data were fitted to the Hill equation using non-linear least-squares regression:
% Inhibition = I_max/[1 + (IC50/[drug])^n]
Where: I_max is the maximal inhibition (constrained to 100% for curve fitting); IC50 is the concentration producing 50% inhibition; [drug] is the compound concentration; n is the Hill coefficient, reflecting binding cooperativity.
Curve fitting was performed using the built-in ‘log(inhibitor) vs. response – Variable slope (four parameters)’ algorithm in Prism. The goodness of fit was assessed using R2 values and visual inspection of residual plots. 95% confidence intervals (CI) for IC50 values were determined using the profile likelihood method. Statistical analysis: IC50 values and Hill coefficients are reported as mean ± standard error of the mean (SEM) from n ≥ 3 independent cells. State-dependent selectivity was calculated as the ratio IC50(rested)/IC50(inactivated). Statistical significance of differences between compound 8 and lidocaine was assessed using unpaired two-tailed Student’s t-test, with p < 0.05 considered statistically significant. Comparisons meeting this criterion are indicated with asterisks in figures: *p < 0.05, **p < 0.01. All data handling and statistical analyses were performed in accordance with Good Laboratory Practice (GLP) guidelines, with raw data archived for audit purposes.
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
- Supporting information for this article is available online at https://doi.org/10.1055/a-2771-1646.
- Supporting Information (PDF) (opens in new window)
-
References and Notes
- 1 Becker DE, Reed KL. Anesth. Prog. 2006; 53: 98
- 2 Becker DE, Reed KL. Anesth. Prog. 2012; 59: 90
- 3 Wang CF, Pancaro C, Gerner P, Strichartz G. Anesthesiology 2011; 114: 135
- 4 Lirk P, Hollmann MW, Strichartz G. Anesth. Analg. 2018; 126: 1381
- 5 Ma H, Pan Z, Lai B, Zan C, Liu H. Drug Des., Dev. Ther. 2023; 30: 2639
- 6 Wei Y, Wu Y, Wen K, Bazybek N, Ma G. J. Mater. Chem. B 2020; 8: 6322
- 7 He Y, Qin L, Huang Y, Ma G. Nanoscale Res. Lett. 2020; 15: 13
- 8 Khabazzadeh H, Saidi K, Seyedi N. J. Chem. Sci. 2009; 121: 429
- 9 Lei H, Jiang N, Miao X, Xing L, Guo M, Liu Y, Xu H, Gong P, Zuo D, Zhai X. Eur. J. Med. Chem. 2019; 171: 297
- 10 Zhang MZ, Chen Q, Yang GF. Eur. J. Med. Chem. 2015; 89: 421
- 11 Connolly DJ, Cusack D, O’Sullivan TP, Guiry PJ. Tetrahedron 2005; 61: 10153
- 12 Catterall WA, Wisedchaisri G, Zheng N. Nat. Chem. Biol. 2017; 13: 455
- 13 Patel BJ, Surana P, Patel KJ. Cureus 2023; 15: e35947
- 14 de Araújo DR, Ribeiro LN. M, de Paula E. Expert Opin. Drug Delivery 2019; 16: 701
- 15 Piegeler T, Votta-Velis EG, Liu G, Place AT, Schwartz DE, Beck-Shimmer B, Minshall RD, Borgeat A. Anesthesiology 2012; 117: 548
- 16 Chamaraux-Tran TN, Piegeler T. Front. Med. 2017; 4: 235
- 17 Bitgen N, Önder G. Ö, Gergin Ö. Ö, Baran M, Gökahmetoğlu G, Yay A. Cukurova Med. J. 2023; 48: 161
- 18 Johnson SM, Saint John BE, Dine AP. Surg. Infect. (Larchmt) 2008; 9: 205
- 19 Hermanns H, Hollmann MW, Stevens MF, Lirk P, Brandenburger T, Piegeler T, Werdehausen R. Br. J. Anaesth. 2019; 123: 335
- 20 Staab HA. Angew. Chem., Int. Ed. Engl. 1962; 1: 351
Corresponding Authors
Publication History
Received: 12 November 2025
Accepted after revision: 10 December 2025
Article published online:
22 January 2026
© 2026. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References and Notes
- 1 Becker DE, Reed KL. Anesth. Prog. 2006; 53: 98
- 2 Becker DE, Reed KL. Anesth. Prog. 2012; 59: 90
- 3 Wang CF, Pancaro C, Gerner P, Strichartz G. Anesthesiology 2011; 114: 135
- 4 Lirk P, Hollmann MW, Strichartz G. Anesth. Analg. 2018; 126: 1381
- 5 Ma H, Pan Z, Lai B, Zan C, Liu H. Drug Des., Dev. Ther. 2023; 30: 2639
- 6 Wei Y, Wu Y, Wen K, Bazybek N, Ma G. J. Mater. Chem. B 2020; 8: 6322
- 7 He Y, Qin L, Huang Y, Ma G. Nanoscale Res. Lett. 2020; 15: 13
- 8 Khabazzadeh H, Saidi K, Seyedi N. J. Chem. Sci. 2009; 121: 429
- 9 Lei H, Jiang N, Miao X, Xing L, Guo M, Liu Y, Xu H, Gong P, Zuo D, Zhai X. Eur. J. Med. Chem. 2019; 171: 297
- 10 Zhang MZ, Chen Q, Yang GF. Eur. J. Med. Chem. 2015; 89: 421
- 11 Connolly DJ, Cusack D, O’Sullivan TP, Guiry PJ. Tetrahedron 2005; 61: 10153
- 12 Catterall WA, Wisedchaisri G, Zheng N. Nat. Chem. Biol. 2017; 13: 455
- 13 Patel BJ, Surana P, Patel KJ. Cureus 2023; 15: e35947
- 14 de Araújo DR, Ribeiro LN. M, de Paula E. Expert Opin. Drug Delivery 2019; 16: 701
- 15 Piegeler T, Votta-Velis EG, Liu G, Place AT, Schwartz DE, Beck-Shimmer B, Minshall RD, Borgeat A. Anesthesiology 2012; 117: 548
- 16 Chamaraux-Tran TN, Piegeler T. Front. Med. 2017; 4: 235
- 17 Bitgen N, Önder G. Ö, Gergin Ö. Ö, Baran M, Gökahmetoğlu G, Yay A. Cukurova Med. J. 2023; 48: 161
- 18 Johnson SM, Saint John BE, Dine AP. Surg. Infect. (Larchmt) 2008; 9: 205
- 19 Hermanns H, Hollmann MW, Stevens MF, Lirk P, Brandenburger T, Piegeler T, Werdehausen R. Br. J. Anaesth. 2019; 123: 335
- 20 Staab HA. Angew. Chem., Int. Ed. Engl. 1962; 1: 351