

Subscribe to RSS
DOI: 10.1055/s-0035-1568597
Elucidation of the Intestinal Absorption Mechanism of Celastrol Using the Caco-2 Cell Transwell Model
Authors
Correspondence
Publication History
received 26 September 2015
revised 01 April 2016
accepted 05 April 2016
Publication Date:
09 May 2016 (online)

Abstract
Celastrol, a triterpenoid isolated from stem (caulis) of Celastrus orbiculatus Thunb. (Celastraceae), has been known to have various pharmacological effects, including anti-inflammatory, anticancer, and antioxidant activities. However, the mechanism of the intestinal absorption of celastrol is unknown. The aim of this study was to investigate the intestinal absorption of celastrol using the Caco-2 cell transwell model. First, the bidirectional transport of celastrol in Caco-2 cell monolayers was observed. Then, the effects of time, concentration, temperature, paracellular pathway, and efflux transport inhibition on the transport of celastrol across the Caco-2 cell monolayers were investigated. The P-glycoprotein inhibitor verapamil and cyclosporin A, the multidrug resistance protein 2 inhibitor MK571, and the breast cancer resistance protein inhibitor reserpine were used. Additionally, the effects of celastrol on the activity of P-glycoprotein were evaluated using the rhodamine 123 uptake assay. In this study, we found that the intestinal transport of celastrol was a time- and concentration-dependent active transport. The paracellular pathway was not involved in the transport of celastrol, and the efflux of celastrol was energy dependent. The results indicated that celastrol is a substrate of P-glycoprotein but not multidrug resistance protein 2 or the breast cancer resistance protein. In addition, celastrol could not affect the uptake of rhodamine 123 in Caco-2 cells, which indicated that celastrol could not inhibit or induce the activity of P-glycoprotein.
Introduction
Celastrol ([Fig. 1]), also known as tripterine, is an active ingredient that was originally identified and extracted from stem (caulis) of Celastrus orbiculatus Thunb. (Celastraceae) [1]. Celastrol has been found to exhibit anti-inflammatory and anticancer properties [2], [3]. Celastrol has exhibited significant cytotoxic activity in different cancer models in vitro [4], [5]. Pharmacokinetic investigations indicated that celastrol is poorly absorbed and the bioavailability of celastrol is low [6], [7], [8]. The metabolic profile of celastrol has been investigated [9], [10], [11]. However, little information is available regarding the intestinal absorption mechanisms. The clinical application of celastrol is restricted because of its narrow therapeutic range and the severe toxicity to the digestive, reproductive, and hematopoietic systems [12], [13], [14]. Therefore, there is a need to investigate its absorption mechanism.

In the drug discovery process, the understanding of the intestinal absorption characteristics of a new molecular entity (NME) to cross biological membranes, particularly the human intestinal mucosa, is critical for clinical development and eventual regulatory approval. Because human intestinal permeability studies are both costly and difficult [15], studies in the human colon carcinoma cell line, Caco-2, are the most commonly used biological tool for screening the intestinal permeability of NMEs during drug discovery and development [16], [17]. Caco-2 cells share similar characteristics with human small intestinal epithelial cells, and this cell line has become a well-accepted model for evaluating intestinal absorption in humans [18], [19]. More importantly, transporter proteins such as P-glycoprotein (P-gp), multidrug resistance protein 1/2 (MRP1/2), and the breast cancer resistance protein (BCRP) are reported to be functionally expressed in Caco-2 cell monolayers [20]. However, the Caco-2 cell transwell model might provide an inaccurate estimation on investigations of intestinal absorption due to their undesirable phenotype and functionality, and a three-dimensional culture model of the intestinal mucosa was developed to study drug absorption, which would improve the estimation accuracy of drug intestinal absorption [21]. Although there are some defects, measurements in the Caco-2 cell line are also accepted by the FDA as a surrogate for human intestinal permeability measurements as part of their Biopharmaceutics Classification System guidance [22]. Therefore, the intestinal transport mechanism of celastrol was investigated using the Caco-2 cell model.
The intestinal tract is the main site of food as well as drug digestion and absorption [23], [24]. Drugs entering into the intestinal epithelial cells might also be secreted back to the intestinal tract [25], [26]. Efflux transporters expressed in the intestinal epithelial cells play an important role in the intestinal absorption of drugs [27], [28]. However, whether or not intestinal efflux transporters are involved in the transport of celastrol is unknown. To the best of our knowledge, there have been little data available concerning the interaction between celastrol and intestinal efflux transporters until now. Therefore, in the present study, the intestinal absorption of celastrol was investigated using the Caco-2 cell transwell model, and the interactions of celastrol with efflux transporters were also elucidated.
Results and Discussion
Before the experiments, celastrol cellular toxicity was examined by an MTT assay, and the results showed that 98 % of cells survived with treatment of 20 µM celastrol; with treatment of 50 µM quercetin, only 65 % of cells survived. The transepithelial electrical resistance (TEER) values were 447 ± 22 Ω · cm2 before the experiments, and the TEER values were also tested after the experiments (422 ± 17 Ω · cm2), which indicated that the Caco-2 cell monolayers were intact before and after the experiments.
To investigate the intestinal transport mechanism of celastrol, a bidirectional transport assay in Caco-2 cell monolayers was preferred. To validate the efflux activity of P-gp, a typical P-gp substrate, digoxin, was used. The efflux ratio of digoxin of 14.5 indicated the abrogation in the presence of a typical P-gp inhibitor, verapamil, which suggested that the efflux activity of P-gp was qualified for the experiment.
[Table 1] shows the permeability of celastrol across Caco-2 cell monolayers. The apparent permeability coefficient (Papp) values of celastrol from the apical-to-basal (AP-BL) side were 3.2 × 10−6 cm/s, 3.5 × 10−6 cm/s, and 4.0 × 10−6 cm/s at 2 µM, 10 µM, and 20 µM celastrol, respectively, indicating that celastrol had passive diffusion ability. However, the basal-to-apical (BL-AP) Papp values of celastrol were much higher than the AP-BL permeability. The calculated efflux ratios of celastrol were 2.9, 3.1, and 2.9 at 2 µM, 10 µM, and 20 µM celastrol, respectively. Intestinal efflux transporters might be involved in the transport of celastrol because the efflux ratio was ≥ 2; however, the specific efflux transporters that were involved in the transport of celastrol is unknown. The transport of celastrol in both directions increased gradually with time and concentration, which is shown in [Fig. 2]. The results showed that the parameters of time and concentration were the important factors influencing the transport of celastrol.

|
Celastrol (µM) |
Papp (10−6 cm/s) |
Efflux ratio |
|
|---|---|---|---|
|
AP-BL |
BL-AP |
||
|
2 |
3.2 ± 0.8 |
9.2 ± 1.2 |
2.9 |
|
10 |
3.5 ± 1.1 |
10.9 ± 2.2 |
3.1 |
|
20 |
4.0 ± 0.9 |
11.5 ± 2.6 |
2.9 |
The effect of the opening of cell junctions on the permeability of a compound is used as a criterion to determine the participation of the paracellular pathway. If the permeability of a solute increases significantly when cell junctions are opened, it is accepted that the paracellular pathway is important for the transport of that solute.
In this study, the paracellular transport study was conducted by modifying the cell junctions by adding EDTA (5 mM), a calcium chelator. Treatment with EDTA produces a substantial opening of cell junctions, as indicated by the increase in the Papp of Lucifer yellow and the decrease in the TEER values.
[Fig. 3] shows the Papp values of celastrol in the AP-BL and BL-AP directions for the Caco-2 monolayers with or without prior treatment with EDTA. The opening of junctions brought little influence in celastrol permeability in either direction (AP-BL: 4.2 × 10−6 cm/s; BL-AP: 11.5 × 10−6 cm/s). These results show that the paracellular pathway was not involved in the transport of celastrol.

A decrease in temperature reduces cellular metabolism and acts as an inhibitor of energy-dependent transport. Therefore, a decrease in the Papp at 4 °C with respect to the Papp at 37 °C might be an indication of participation of this type of transport. The permeability coefficient in the AP-BL direction obtained at 4 °C was (3.3 ± 0.4) × 10− 6 cm/s, similar to the values obtained at 37 °C. This finding indicated that there was no or only minor energy-dependent transport in the process of absorption of this element.
In the BL-AP direction, there was a significant decrease in transport at the low temperature [Papp (cm/s): 4 °C: (6.4 ± 0.7) × 10−6; 37 °C: (10.9 ± 1.2) × 10−6], indicating the possible participation of temperature-dependent transport in the secretory direction.
Efflux transporters commonly expressed in the intestinal membranes include P-gp, MRP2, and BCRP. These proteins are members of the ATP-binding cassette (ABC) transport proteins that use ATP as an energy source to transport substrates against a concentration gradient from the cytoplasm of intestinal cells back to the intestinal lumen or to the blood [29], [30]. As shown in [Fig. 4], in the presence of MK571 or reserpine, there was almost no effect on Papp(AP-BL) and Papp(BL-AP), which indicates that celastrol was not a substrate of MRP2 and BCRP. However, in the presence of the P-gp inhibitors verapamil and cyclosporin A, the Papp(AP-BL) was significantly increased as the Papp(BL-AP) decreased, and the efflux ratio of celastrol decreased from 4.5 to 1.3 and 1.0. These results suggest that celastrol is a substrate of P-gp. Therefore, it could be speculated that celastrol might first be absorbed into the intestinal epithelial cells, and then P-gp expressed in the intestine might pump out celastrol from the intestinal epithelial cells and limit the absorption of celastrol.
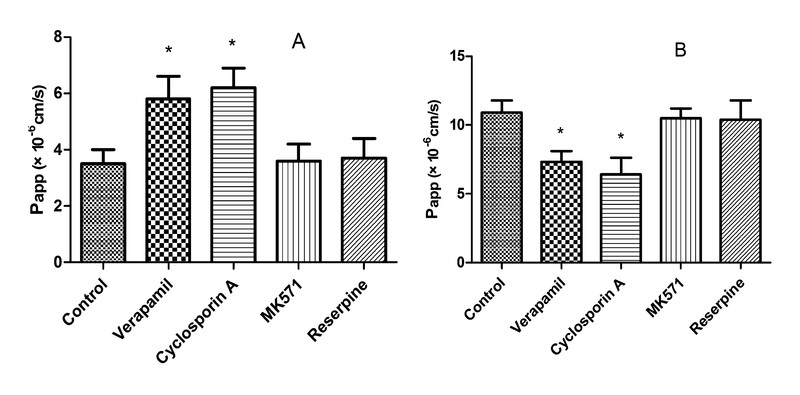
The efflux transporter P-gp is an important biological barrier against foreign agents because of its high expression in the gastrointestinal tract [31]. Therefore, this protein serves as a major determinant of drug absorption in the small intestine. Numerous natural compounds from medicinal herbs have been reported to be inhibitors of P-gp, and these compounds could enhance the oral bioavailability of P-gp substrates and also reverse multidrug resistance induced by chemotherapeutic agents [32], [33]. There are several reports on the pharmacokinetic drug-drug interactions based on the inhibition or induction of transporters [34], [35]; therefore, the effects of celastrol on the activity of P-gp should be elucidated.
[Fig. 5] shows the accumulation of rhodamine 123 in the Caco-2 cell in the presence of 50 µM celastrol. Verapamil and cyclosporin A, known inhibitors of P-gp, increased the cellular accumulation of rhodamine 123. However, celastrol could not increase the cellular accumulation of rhodamine 123, which indicates that celastrol could not affect the P-gp-mediated efflux of rhodamine 123 and had little influence on the activity of P-gp.

Celastrol has been used for different diseases and might be used in combination with other drugs. The coadministration of celastrol with P-gp inhibitors might increase the bioavailability of celastrol and increase its toxic effect in vivo. Therefore, drug-drug interactions might occur when celastrol is coadministered with P-gp inhibitors.
This study had some limitations. First, celastrol is an acid compound, and the study could provide more details if the deprotonated form of celastrol in solution (pH 7.4) was used. In future research, the pKa value of celastrol and the deprotonated form of celastrol in solution (pH 7.4) should be investigated. Second, the effects of celastrol on the activity of other membrane transporters were not investigated in this study; our future research will focus on this subject.
In this study, the intestinal absorption mechanism of celastrol was investigated using the Caco-2 cell monolayer model. The results indicated that the intestinal transport of celastrol occurred in a time- and concentration-dependent passive transport mechanism, and the efflux of celastrol was an energy-dependent transport. P-gp was involved in the transport of celastrol, which hindered the absorption of celastrol in the intestine, and celastrol had little effect on the activity of P-gp. We hope this research will be helpful for the development of celastrol as a chemotherapeutic agent.
Materials and Methods
Drugs and reagents
Celastrol (> 98 %), hydrocortisone (> 98 %), reserpine (> 98 %), quercetin (> 98 %), and verapamil (> 98 %) were purchased from the National Institutes for Food and Drug Control (Beijing, China). Cyclosporin A (> 99 %) was purchased from Gene Operation. MK-571 (> 95 %) and rhodamine 123 were purchased from Sigma-Aldrich. DMEM, FBS, and trypsin were obtained from Gibico. Methanol and acetonitrile were obtained from J&K. Other reagents were of analytical grade or better.
Cell culture
The Caco-2 cell line was obtained from the American Type Culture Collection. The Caco-2 cell transport experiment was performed according to a method previously reported [36]. Briefly, Caco-2 cells were cultured in DMEM high-glucose media containing 15 % FBS, 1 % NEAA, and 100 U/mL of penicillin and streptomycin. The cells were cultured at 37 °C with 5 % CO2. For transport studies, cells at passage 30–40 were seeded on transwell polycarbonate insert filters (1.12 cm2 surface, 0.4 µm pore size, 12 mm diameter; Corning Costar Corporation) in 12-well plates at a density of 1 × 105 cells/cm2. The cells were allowed to grow for 21 days. For the first 7 days, the media was replaced every 2 days, and then the media was replaced daily thereafter. The TEER of the monolayer cells was measured by using Millicell ERS-2 (Millipore Corporation), and the TEER value should exceed 400 Ω · cm2. The integrity of the Caco-2 monolayers was confirmed by the paracellular flux of Lucifer yellow of less than 1 % per hour. The alkaline phosphatase activity was validated using an Alkaline Phosphatase Assay Kit. The efflux activity of P-gp was validated using a typical P-gp substrate, digoxin (25 µM). The qualified monolayers were used for the transport studies.
Cell viability assay
Before the experiments, celastrol cellular toxicity was examined by an MTT assay. Briefly, Caco-2 cells were seeded in 96-well plates at a density of 4000 cells per well. Twenty-four hours later, the cells were treated with increasing concentrations of celastrol (1–100 µM) and cultured for 3 h; quercetin (50 µM) was used as a positive control [37], and blank Hanksʼ balanced salt solution (HBSS) was used as a negative control. At the end of treatment, the MTT uptake method for cell viability determination was conducted according to the manufacturerʼs protocol (Promega), and the plate was read at 490 nm.
Transport studies
Before the transport experiments, the cell monolayers were rinsed twice using warm (37 °C) HBSS, and then the cells were incubated at 37 °C for 20 min. After preincubation, the cell monolayers were incubated with celastrol in fresh incubation media from either the apical (AP) or basolateral (BL) side for the designated time at 37 °C. The volume of incubation media on the AP and BL sides was 0.5 mL and 1.5 mL, respectively, and a 100-µL aliquot of the incubation solution was withdrawn at the designated time points (30, 60, 90, and 120 min) from the receiver compartment and replaced with the same volume of a fresh pre-warmed HBSS buffer; three concentrations of celastrol (2, 10, and 20 µM) were tested.
Paracellular transport of celastrol
The participation of the paracellular pathway in the transport of celastrol was evaluated by modulating the cell junctions in both directions (AP-BL and BL-AP). For this purpose, the cell monolayer was incubated with 5 mM EDTA in PBS free of Ca2+ and Mg2+ for 5 min. Then, the standard of celastrol (10 µM), prepared in medium consisting of 50 % HBSS free of Ca2+ and Mg2+ supplemented with 10 mM HEPES and 50 % HBSS supplemented with 10 mM HEPES, was added to the donor compartment. The acceptor medium was collected at various times (30, 60, 90, and 120 min), and the concentration of celastrol was determined. At the same time, the efficiency of EDTA in modulating the cell junctions was monitored by determining the Papp value of Lucifer yellow before and after the EDTA treatment.
Effects of temperature on transport of celastrol
The study of transport at 37 °C and 4 °C was conducted in the AP-BL and BL-AP directions. For this study, 10 µM celastrol prepared in HBSS supplemented with 10 mM HEPES were added to the donor compartment, and a 100-µL aliquot of the incubation solution was withdrawn at designated time points (30, 60, 90, and 120 min) from the receiver compartment and replaced with the same volume of fresh pre-warmed HBSS buffer.
Effect of several transporter inhibitors on the transport of celastrol
The inhibitory effects of transporter inhibitors on celastrol flux by Caco-2 cells were investigated by adding 50 µM verapamil, MK-571, reserpine, and 10 µM cyclosporin A to both sides of the cell monolayers. The permeability of celastrol (10 µM) in the above conditions in both directions (A–B and B-A) was measured after incubation for 30, 60, 90, and 120 min at 37 °C.
Measurement of cellular accumulation of rhodamine 123 in Caco-2 cells
The accumulation of rhodamine 123, a fluorescent substrate of P-gp, was measured, and the effects of celastrol were determined as described previously [38]. Briefly, Caco-2 cells, plated at 1 × 105 cells/well in 24-well plates, were incubated with 20 µM rhodamine 123 in the absence or presence of celastrol (50 µM) for 1 h in a CO2 incubator at 37 °C. After the incubation, the medium was removed by aspiration and the cells were washed with ice-cold PBS and lysed with 0.1 % triton-X100 in PBS. The fluorescence intensity was measured with a microplate fluorometer (Fluoroskan Ascent). The excitation and emission wavelengths were 485 and 538 nm, respectively. The protein concentrations were measured by the Lowry method using a Bio-Rad DC protein assay kit (Bio-Rad Laboratories) with bovine serum albumin as the standard. The accumulation ratios were calculated using the accumulation of rhodamine 123 in cells incubated without celastrol as a control.
Determination of celastrol using HPLC-UV
The celastrol concentration was analyzed by HPLC-UV according to a reported method with some modification [39]. An Agilent HPLC 1200 system was used for the determination. Samples were separated with a Shiseido C18 column (3.0 × 100 mm, 3 µm). The mobile phase consisted of 80 % acetonitrile and 20 % formic acid (0.1 %) in water. The column temperature was set at 30 °C, and the flow rate was set at 0.8 mL/min. Celastrol was detected at a wavelength of 456 nm. The injection volume was 20 µL.
The calibration curve (Y = 0.287 X + 2.618) was linear over the range of 15–15 000 ng/mL with a correlation coefficient of 0.999. The limit of quantification and the limit of detection of this method were 15 and 5 ng/mL, respectively. The intra- and interday precision values (RSD) were less than 10 %, and the accuracy (RE) ranged from − 6.65 % to 8.42 %. These data indicate that the accuracy and precision of the method were satisfactory.
Data analysis
The apparent permeability coefficient (Papp) was calculated using the equation of Artursson and Karlsson [40]: Papp = (ΔQ/Δt) × [1/(A × C0)], where Papp is the apparent permeability coefficient (cm/s), ΔQ/Δt (µmol/s) is the rate at which the compound appears at the receiver chamber, C0 (µmol/L) is the initial concentration of the compound in the donor chamber, and A (cm2) represents the surface area of the cell monolayer. Data were collected from the three separate experiments, and each experiment was performed in triplicate. All results are expressed as the mean ± SD. Group comparisons were conducted using the unpaired t-test. Differences between groups were considered statistically significant for p values < 0.05.
Acknowledgements
This study was supported by the National Nature Science Foundation of China (30700387).
Conflict of Interest
The authors declare no conflict of interest.
* The first three authors contributed equally to this work.
-
References
- 1 Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 2006; 66: 4758-4765
- 2 Dai Y, Desano J, Tang W, Meng X, Meng Y, Burstein E, Lawrence TS, Xu L. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One 2010; 5: e14153
- 3 Ji N, Li J, Wei Z, Kong F, Jin H, Chen X, Li Y, Deng Y. Effect of celastrol on growth inhibition of prostate cancer cells through the regulation of hERG channel in vitro . Biomed Res Int 2015; 2015: 308475
- 4 Nabekura T, Hiroi T, Kawasaki T, Uwai Y. Effects of natural nuclear factor-kappa B inhibitors on anticancer drug efflux transporter human P-glycoprotein. Biomed Pharmacother 2015; 70: 140-145
- 5 Shrivastava S, Jeengar MK, Reddy VS, Reddy GB, Naidu VG. Anticancer effect of celastrol on human triple negative breast cancer: possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3 K/Akt pathways. Exp Mol Pathol 2015; 98: 313-327
- 6 Chen Y, Yuan L, Zhou L, Zhang ZH, Cao W, Wu Q. Effect of cell-penetrating peptide-coated nanostructured lipid carriers on the oral absorption of tripterine. Int J Nanomedicine 2012; 7: 4581-4591
- 7 Qi X, Qin J, Ma N, Chou X, Wu Z. Solid self-microemulsifying dispersible tablets of celastrol: formulation development, charaterization and bioavailability evaluation. Int J Pharm 2014; 472: 40-47
- 8 Zhang J, Li CY, Xu MJ, Wu T, Chu JH, Liu SJ, Ju WZ. Oral bioavailability and gender-related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats. J Ethnopharmacol 2012; 144: 195-200
- 9 Jin C, He X, Zhang F, He L, Chen J, Wang L, An L, Fan Y. Inhibitory mechanisms of celastrol on human liver cytochrome P450 1A2, 2C19, 2D6, 2E1 and 3A4. Xenobiotica 2015; 45: 571-577
- 10 Sun M, Tang Y, Ding T, Liu M, Wang X. Inhibitory effects of celastrol on rat liver cytochrome P450 1A2, 2C11, 2D6, 2E1 and 3A2 activity. Fitoterapia 2014; 92: 1-8
- 11 Yang H, Landis-Piwowar KR, Chen D, Milacic V, Dou QP. Natural compounds with proteasome inhibitory activity for cancer prevention and treatment. Curr Protein Pept Sci 2008; 9: 227-239
- 12 Pinna GF, Fiorucci M, Reimund JM, Taquet N, Arondel Y, Muller CD. Celastrol inhibits pro-inflammatory cytokine secretion in Crohnʼs disease biopsies. Biochem Biophys Res Commun 2004; 322: 778-786
- 13 Sharma S, Mishra R, Walker BL, Deshmukh S, Zampino M, Patel J, Anamalai M, Simpson D, Singh IS, Kaushal S. Celastrol, an oral heat shock activator, ameliorates multiple animal disease models of cell death. Cell Stress Chaperones 2015; 20: 185-201
- 14 Wang S, Liu K, Wang X, He Q, Chen X. Toxic effects of celastrol on embryonic development of zebrafish (Danio rerio). Drug Chem Toxicol 2011; 34: 61-65
- 15 Lennernäs H. Intestinal permeability and its relevance for absorption and elimination. Xenobiotica 2007; 37: 1015-1051
- 16 Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev 2001; 46: 27-43
- 17 Hua WJ, Fang HJ, Hua WX. Transepithelial transport of rosuvastatin and effect of ursolic acid on its transport in Caco-2 monolayers. Eur J Drug Metab Pharmacokinet 2012; 37: 225-231
- 18 Artursson P. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorptive (Caco-2) cells. J Pharm Sci 1990; 79: 476-482
- 19 Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun 1991; 175: 880-885
- 20 Gutmann H, Fricker G, Torok M, Michael S, Beglinger C, Drewe J. Evidence for different ABC-transporters in Caco-2 cells modulating drug uptake. Pharm Res 1999; 16: 402-407
- 21 Li N, Wang D, Sui Z, Qi X, Ji L, Wang X, Yang L. Development of an improved three-dimensional in vitro intestinal mucosa model for drug absorption evaluation. Tissue Eng Part C Methods 2013; 19: 708-719
- 22 Larregieu CA, Benet LZ. Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco-2 as a surrogate for human intestinal permeability measurements. AAPS J 2013; 15: 483-497
- 23 Chen Y, Wang Y, Zhou J, Gao X, Qu D, Liu C. Study on the mechanism of intestinal absorption of epimedins A, B and C in the Caco-2 cell model. Molecules 2014; 19: 686-698
- 24 Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989; 96: 736-749
- 25 Joyce H, McCann A, Clynes M, Larkin A. Influence of multidrug resistance and drug transport proteins on chemotherapy drug metabolism. Expert Opin Drug Metab Toxicol 2015; 11: 795-809
- 26 Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest 1998; 101: 289-294
- 27 Lee WC, Peng CC, Chang CH, Huang SH, Chyau CC. Extraction of antioxidant components from Bidens pilosa flowers and their uptake by human intestinal Caco-2 cells. Molecules 2013; 18: 1582-1601
- 28 Nguyen MA, Staubach P, Wolffram S, Langguth P. Effect of single-dose and short-term administration of quercetin on the pharmacokinetics of talinolol in humans – Implications for the evaluation of transporter-mediated flavonoid-drug interactions. Eur J Pharm Sci 2014; 61: 54-60
- 29 Awortwe C, Fasinu PS, Rosenkranz B. Application of Caco-2 cell line in herb-drug interaction studies: current approaches and challenges. J Pharm Pharm Sci 2014; 17: 1-19
- 30 Manda VK, Avula B, Ali Z, Khan IA, Walker LA, Khan SI. Evaluation of in vitro absorption, distribution, metabolism, and excretion (ADME) properties of mitragynine, 7-hydroxymitragynine, and mitraphylline. Planta Med 2014; 80: 568-576
- 31 Zhou S, Lim LY, Chowbay B. Herbal modulation of P-glycoprotein. Drug Metab Rev 2004; 36: 57-104
- 32 Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013; 7: 27-34
- 33 Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets 2013; 13: 326-346
- 34 Yang JM, Ip SP, Xian Y, Zhao M, Lin ZX, Yeung JH, Chan RC, Lee SS, Che CT. Impact of the herbal medicine Sophora flavescens on the oral pharmacokinetics of indinavir in rats: the involvement of CYP3 A and P-glycoprotein. PLoS One 2012; 7: e31312
- 35 Zhang Y, Li J, Lei X, Zhang T, Liu G, Yang M, Liu M. Influence of verapamil on pharmacokinetics of triptolide in rats. Eur J Drug Metab Pharmacokinet advance online publication 9 April 2015
- 36 Li N, Tsao R, Sui Z, Ma J, Liu Z. Intestinal transport of pure diester-type alkaloids from an aconite extract across the Caco-2 cell monolayer model. Planta Med 2012; 78: 692-697
- 37 Zhang XA, Zhang S, Yin Q, Zhang J. Quercetin induces human colon cancer cells apoptosis by inhibiting the nuclear factor-kappa B pathway. Pharmacogn Mag 2015; 11: 404-409
- 38 Liang Y, Zhou Y, Zhang J, Liu Y, Guan T, Wang Y, Xing L, Rao T, Zhou L, Hao K, Xie L, Wang GJ. In vitro to in vivo evidence of the inhibitor characteristics of Schisandra lignans toward P-glycoprotein. Phytomedicine 2013; 20: 1030-1038
- 39 Sun JN, Shi YP, Chen J. Ultrasound-assisted ionic liquid dispersive liquid-liquid microextraction coupled with high performance liquid chromatography for sensitive determination of trace celastrol in urine. J Chromatogr B Analyt Technol Biomed Life Sci 2011; 879: 3429-3433
- 40 Manda VK, Avula B, Ali Z, Wong YH, Smillie TJ, Khan IA, Khan SI. Characterization of in vitro ADME properties of diosgenin and dioscin from Dioscorea villosa . Planta Med 2013; 79: 1421-1428
Correspondence
-
References
- 1 Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 2006; 66: 4758-4765
- 2 Dai Y, Desano J, Tang W, Meng X, Meng Y, Burstein E, Lawrence TS, Xu L. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One 2010; 5: e14153
- 3 Ji N, Li J, Wei Z, Kong F, Jin H, Chen X, Li Y, Deng Y. Effect of celastrol on growth inhibition of prostate cancer cells through the regulation of hERG channel in vitro . Biomed Res Int 2015; 2015: 308475
- 4 Nabekura T, Hiroi T, Kawasaki T, Uwai Y. Effects of natural nuclear factor-kappa B inhibitors on anticancer drug efflux transporter human P-glycoprotein. Biomed Pharmacother 2015; 70: 140-145
- 5 Shrivastava S, Jeengar MK, Reddy VS, Reddy GB, Naidu VG. Anticancer effect of celastrol on human triple negative breast cancer: possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3 K/Akt pathways. Exp Mol Pathol 2015; 98: 313-327
- 6 Chen Y, Yuan L, Zhou L, Zhang ZH, Cao W, Wu Q. Effect of cell-penetrating peptide-coated nanostructured lipid carriers on the oral absorption of tripterine. Int J Nanomedicine 2012; 7: 4581-4591
- 7 Qi X, Qin J, Ma N, Chou X, Wu Z. Solid self-microemulsifying dispersible tablets of celastrol: formulation development, charaterization and bioavailability evaluation. Int J Pharm 2014; 472: 40-47
- 8 Zhang J, Li CY, Xu MJ, Wu T, Chu JH, Liu SJ, Ju WZ. Oral bioavailability and gender-related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats. J Ethnopharmacol 2012; 144: 195-200
- 9 Jin C, He X, Zhang F, He L, Chen J, Wang L, An L, Fan Y. Inhibitory mechanisms of celastrol on human liver cytochrome P450 1A2, 2C19, 2D6, 2E1 and 3A4. Xenobiotica 2015; 45: 571-577
- 10 Sun M, Tang Y, Ding T, Liu M, Wang X. Inhibitory effects of celastrol on rat liver cytochrome P450 1A2, 2C11, 2D6, 2E1 and 3A2 activity. Fitoterapia 2014; 92: 1-8
- 11 Yang H, Landis-Piwowar KR, Chen D, Milacic V, Dou QP. Natural compounds with proteasome inhibitory activity for cancer prevention and treatment. Curr Protein Pept Sci 2008; 9: 227-239
- 12 Pinna GF, Fiorucci M, Reimund JM, Taquet N, Arondel Y, Muller CD. Celastrol inhibits pro-inflammatory cytokine secretion in Crohnʼs disease biopsies. Biochem Biophys Res Commun 2004; 322: 778-786
- 13 Sharma S, Mishra R, Walker BL, Deshmukh S, Zampino M, Patel J, Anamalai M, Simpson D, Singh IS, Kaushal S. Celastrol, an oral heat shock activator, ameliorates multiple animal disease models of cell death. Cell Stress Chaperones 2015; 20: 185-201
- 14 Wang S, Liu K, Wang X, He Q, Chen X. Toxic effects of celastrol on embryonic development of zebrafish (Danio rerio). Drug Chem Toxicol 2011; 34: 61-65
- 15 Lennernäs H. Intestinal permeability and its relevance for absorption and elimination. Xenobiotica 2007; 37: 1015-1051
- 16 Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev 2001; 46: 27-43
- 17 Hua WJ, Fang HJ, Hua WX. Transepithelial transport of rosuvastatin and effect of ursolic acid on its transport in Caco-2 monolayers. Eur J Drug Metab Pharmacokinet 2012; 37: 225-231
- 18 Artursson P. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorptive (Caco-2) cells. J Pharm Sci 1990; 79: 476-482
- 19 Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun 1991; 175: 880-885
- 20 Gutmann H, Fricker G, Torok M, Michael S, Beglinger C, Drewe J. Evidence for different ABC-transporters in Caco-2 cells modulating drug uptake. Pharm Res 1999; 16: 402-407
- 21 Li N, Wang D, Sui Z, Qi X, Ji L, Wang X, Yang L. Development of an improved three-dimensional in vitro intestinal mucosa model for drug absorption evaluation. Tissue Eng Part C Methods 2013; 19: 708-719
- 22 Larregieu CA, Benet LZ. Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco-2 as a surrogate for human intestinal permeability measurements. AAPS J 2013; 15: 483-497
- 23 Chen Y, Wang Y, Zhou J, Gao X, Qu D, Liu C. Study on the mechanism of intestinal absorption of epimedins A, B and C in the Caco-2 cell model. Molecules 2014; 19: 686-698
- 24 Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989; 96: 736-749
- 25 Joyce H, McCann A, Clynes M, Larkin A. Influence of multidrug resistance and drug transport proteins on chemotherapy drug metabolism. Expert Opin Drug Metab Toxicol 2015; 11: 795-809
- 26 Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest 1998; 101: 289-294
- 27 Lee WC, Peng CC, Chang CH, Huang SH, Chyau CC. Extraction of antioxidant components from Bidens pilosa flowers and their uptake by human intestinal Caco-2 cells. Molecules 2013; 18: 1582-1601
- 28 Nguyen MA, Staubach P, Wolffram S, Langguth P. Effect of single-dose and short-term administration of quercetin on the pharmacokinetics of talinolol in humans – Implications for the evaluation of transporter-mediated flavonoid-drug interactions. Eur J Pharm Sci 2014; 61: 54-60
- 29 Awortwe C, Fasinu PS, Rosenkranz B. Application of Caco-2 cell line in herb-drug interaction studies: current approaches and challenges. J Pharm Pharm Sci 2014; 17: 1-19
- 30 Manda VK, Avula B, Ali Z, Khan IA, Walker LA, Khan SI. Evaluation of in vitro absorption, distribution, metabolism, and excretion (ADME) properties of mitragynine, 7-hydroxymitragynine, and mitraphylline. Planta Med 2014; 80: 568-576
- 31 Zhou S, Lim LY, Chowbay B. Herbal modulation of P-glycoprotein. Drug Metab Rev 2004; 36: 57-104
- 32 Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013; 7: 27-34
- 33 Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets 2013; 13: 326-346
- 34 Yang JM, Ip SP, Xian Y, Zhao M, Lin ZX, Yeung JH, Chan RC, Lee SS, Che CT. Impact of the herbal medicine Sophora flavescens on the oral pharmacokinetics of indinavir in rats: the involvement of CYP3 A and P-glycoprotein. PLoS One 2012; 7: e31312
- 35 Zhang Y, Li J, Lei X, Zhang T, Liu G, Yang M, Liu M. Influence of verapamil on pharmacokinetics of triptolide in rats. Eur J Drug Metab Pharmacokinet advance online publication 9 April 2015
- 36 Li N, Tsao R, Sui Z, Ma J, Liu Z. Intestinal transport of pure diester-type alkaloids from an aconite extract across the Caco-2 cell monolayer model. Planta Med 2012; 78: 692-697
- 37 Zhang XA, Zhang S, Yin Q, Zhang J. Quercetin induces human colon cancer cells apoptosis by inhibiting the nuclear factor-kappa B pathway. Pharmacogn Mag 2015; 11: 404-409
- 38 Liang Y, Zhou Y, Zhang J, Liu Y, Guan T, Wang Y, Xing L, Rao T, Zhou L, Hao K, Xie L, Wang GJ. In vitro to in vivo evidence of the inhibitor characteristics of Schisandra lignans toward P-glycoprotein. Phytomedicine 2013; 20: 1030-1038
- 39 Sun JN, Shi YP, Chen J. Ultrasound-assisted ionic liquid dispersive liquid-liquid microextraction coupled with high performance liquid chromatography for sensitive determination of trace celastrol in urine. J Chromatogr B Analyt Technol Biomed Life Sci 2011; 879: 3429-3433
- 40 Manda VK, Avula B, Ali Z, Wong YH, Smillie TJ, Khan IA, Khan SI. Characterization of in vitro ADME properties of diosgenin and dioscin from Dioscorea villosa . Planta Med 2013; 79: 1421-1428