
RSS-Feed abonnieren
DOI: 10.1055/s-0036-1589500
Recent Advances in the Synthesis of Hydrogenated Azocine-Containing Molecules
Publikationsverlauf
Received: 22. Januar 2017
Accepted after revision: 05. April 2017
Publikationsdatum:
01. August 2017 (online)

Dedicated to the memory of Professor N. S. Prostakov (1917–2007) on the occasion of his 100th anniversary.
Abstract
This review covers recent advances in synthesis of azocine-containing systems. The most approaches towards azocines are discussed.
1 Introduction
2 Ring-Expansion Reaction
2.1 Ring-Expansion Reaction of Cyclopentane Containing the 1,4-Diketone Moiety with Primary Amines (from 5 to 8)
2.2 Ring-Expansion Reaction of Annulated Tetrahydropyridines under the Action of Activated Alkynes (from 6 to 8)
2.3 Reductive Ring-Expansion Reaction of Cyclic Oximes
2.4 Other Ring-Expansion Reactions
3 Heck Reaction
4 Cycloaddition
5 Ring-Closing Metathesis (RCM)
6 Cyclization
6.1 Metal-Catalyzed Cyclization
6.2 Radical Cyclization
6.3 Friedel–Crafts Cyclization
6.4 Other Examples of Cyclizations
7 Microwave- and Photo-Assisted Reactions
8 Other Methods
8.1 Cascade and Tandem Reactions
8.2 Aldol Condensation
8.3 Thermolysis
8.4 Ring Opening
8.5 Other Methods
9 Conclusion
#
Key words
azocine - ring-closing metathesis - cycloaddition - ring expansion - Heck reaction - domino reactionBiographical Sketches

Prof. Leonid G. Voskressensky was born in 1968 in Moscow, Russia. He obtained his B.Sc. in chemistry from the Peoples’ Friendship University of Russia (PFUR) in 1992, and his M.Sc. in 1994. He obtained his Ph.D. in organic chemistry from the same university in 1999. In 2001 he joined the group of Prof. Cosimo Altomare (Universita degli Studi di Bari, Italy) as a postdoctoral fellow in medicinal chemistry. In 2001, he became assistant professor, in 2006 associate professor, and in 2011 full professor in the organic chemistry department at PFUR. Since 2013 he has been the Dean of the Science Faculty at PFUR. His group’s scientific interests focus mainly on domino reaction methodology, new multicomponent reactions, and medicinal chemistry.

Dr. Anna Listratova was born in Moscow, Russia. She obtained her B.Sc. in chemistry from the People’s Friendship University of Russia (PFUR) in 2003, followed by an M.Sc. degree in 2005. She obtained the Ph.D. degree in organic chemistry from the same university in 2008 under guidance of Prof. L. G. Voskressensky and remains a member of his group.
Introduction
The chemistry of annulated azocines has not been explored in detail owing to the lack of efficient methods for their synthesis. The only exception is azocinoindoles, which have been investigated extensively due to the great number of alkaloids with an azocinoindole fragment in their structure. This review highlights most recent approaches towards annulated azocine derivatives published after the year 2009; the previous review was published in 2008.[1]

# 2
Ring-Expansion Reactions
2.1Ring-Expansion Reaction of Cyclopentane Containing the 1,4-Diketone Moiety with Primary Amines (from 5 to 8)


In 2006 the Cristoffers group[2] discovered a novel bismuth-catalyzed ring-expansion reaction of 1,4-diketones with primary amines that furnished an eight-membered ring. In 2011, they extended their relatively simple method to the synthesis of annulated azocines. Thus, starting from ethyl 1-oxo-indane-2-carboxylate 1, containing a 1,4-diketone motif, and primary amines under the bismuth-catalyzed ring-expansion reaction conditions gave benzo[c]azocine derivatives 2 in moderate yields (Scheme [1]).[3] It was also shown that, in some cases, the presence of bismuth nitrate was not essential.[3]
A bismuth-free strategy of ring enlargement was also successful in the case of regioisomeric pyrido[c]azocines.[4] Synthesized from commercially available materials, three cyclopentapyridine derivatives 3, 6, and 9, containing β-oxo ester moieties, were alkylated with phenacyl bromide to give 1,4-diketones 4, 7, and 10. The latter were subjected to ring-expansion reactions with methylamine giving pyrido[2,3-c]azocine 5 (Scheme [2]), pyrido[3,4-c]azocine 8 (Scheme [3]), and pyrido[3,2-c]azocine 11 (Scheme [4]) in 36–64% yield.

In 2015, a six-step sequence for the synthesis of regioisomeric thieno[c]azocines started from commercially available bromothiophenecarboxylic acids was worked out.[5] Isopropyl esters of the bromothiophenecarboxylic acids were subjected to Heck reaction followed by catalytic hydrogenation and Dieckmann condensation giving the cyclic β-oxo esters 12, 15, and 18, alkylation of which with phenacyl bromide led to 1,4-diketones 13, 16, and 19. The following step, a bismuth-catalyzed ring expansion of cyclopentathiophene derivatives 13, 16, and 19 with methylamine, produced the target tetrahydrothieno[3,2-c]azocine 14 (Scheme [5]), tetrahydrothieno[3,4-c]azocine 17 (Scheme [6]), and tetrahydrothieno[2,3-c]azocine 20 (Scheme [7]). Overall yields for the final products were 25%, 16%, and 12%, respectively.



# 2.2
Ring-Expansion Reaction of Annulated Tetrahydropyridines under the Action of Activated Alkynes (from 6 to 8)
In 2002, an alkyne-induced ring-expansion reaction of annulated tetrahydropyridines leading to the formation of azocine rings was found.[6] It is presumed that ring-expansion reaction involves the Michael addition of the tertiary N-atom in the (hetero)annulated pyridine system to the triple bond of the activated alkyne, followed by a nucleophilic substitution (SN) reaction in zwitterionic intermediate A (Scheme [8]).
Over the last 8 years this method was successfully applied to the synthesis of various annulated azocines: triazolopyrimido[4,5-d]azocines 21,[7] [8] tetrahydro[1]benzothieno[3,2-d]azocines 22,[9] hexahydropyrimido[4,5-d]azocines 23 and -[5,4-d]azocines 24,[10] tetrahydropyrimido[4,5-d]azocines 25,[11] tetrahydrobenzofuro[3,2-d]azocine 26,[12] tetrahydrothieno[2,3-d]azocines 27,[13] tetrahydroazocino[5,4-b]indoles 28,[14] [15] hexahydropyrimidothieno[3,2-d]azocines 29,[16] [17] and benzo[d]azocines 30,[18] including systems obtained for the first time, tetrahydrothieno[3,2-d]azocines 31 [19] and tetrahydrochromeno[4,3-d]azocine 32 (Figure [1]).[20]


Attempts to combine the aforesaid ring expansion and Ugi or Ugi-azide transformations into a single multicomponent reaction succeeded and provided thieno[3,2-d]azocine 33 [21] (Scheme [9]) and tetrazolyl-substituted benzo[d]azocine 34 [18] (Scheme [10]) in moderate yields.


# 2.3
Reductive Ring-Expansion Reaction of Cyclic Oximes

Using a method devised by Cho, Tokuyama, and co-workers, regiocontrolled reductive ring-expansion of cyclic oxime with diisobutylaluminum hydride gave benzo[b]azocine 35 and dibenzo[b,f]azocine 36 in high yields as a single regioisomer with the nitrogen atom located in the position neighboring the aromatic ring (Scheme [11]).[22] [23] [24]
Based on this ring-expansion reaction of oximes, a concise synthesis of 17β-HSD3 inhibitor with a dibenzoazocine skeleton was carried out.[25] All attempts to convert oximes 37a,b into a single regioisomer failed, hence a mixture of dibenzoazocines 38a and 39a in the ratio 2:1 or dibenzoazocines 38b and 39b in the ratio 6:1 was used. After acylation of dibenzoazocines 38 and 39 the obtained regioisomers 40 and 41 were separated. Compound 40a was coupled under the Suzuki–Miyaura coupling conditions to provide 17β-HSD3 inhibitor 42 in 70% yield. Desulfurization of 42 with Raney Ni gave also 17β-HSD3 inhibitor 43 (Scheme [12]). Since 17β-hydroxysteroid dehydrogenase type 3 (17βHSD3) is an enzyme involved in testosterone biosynthesis, inhibitors of 17β-HSD3 could provide new medicines for the treatment of prostate cancer.

# 2.4
Other Ring-Expansion Reactions
Treatment of 1-vinyl-substituted indolinium salt 44 in refluxing THF with the Hoveyda–Grubbs second-generation catalyst (H-G II) resulted in allylic rearrangement to give azocino[5,4-b]indole 45, the product of ring expansion, in 44% yield (Scheme [13]).[26]
A conceptually new and elegant strategy for the construction of 1H-azocino[5,4-b]indoles 47 and 48 via a gold-catalyzed ring expansion of 2-propargyl-β-tetrahydrocarbolines 46 was developed by Zhang and co-workers.[27] The azocinoindoles 47 and 48 were obtained in moderate to excellent yields. The method features mild conditions and wide functional group tolerance (Scheme [14]).


Binaphthyl-azocines 50 were synthesized by the direct copper-catalyzed ring-expansion reaction of binaphthyl-azepines 49 and α-diazocarbonyl reagents.[28] This transformation is considered to be an example of a [1,2-]Stevens rearrangement and presents a facile access to binaphthyl-azocines in moderate to high yields (Scheme [15]).

Naphtho[1,8-ef]pyrimido[4,5-b]azocines 53 were prepared from acenaphthoquinone (51) and 6-aminouracil derivatives 52 in high yields.[29] The ring expansion was carried out as a one-pot process and included two steps: the addition reaction of starting compounds and the subsequent oxidation cleavage of the intermediate in the presence of Pd(OAc)4 (Scheme [16]).

Azocine 55 and annulated azocine 57 were produced by a ring-expansion transformation of the piperidine ring of compounds 54 and 56 under the action of methyl propynoate and dimethyl butynedioate, respectively (Scheme [17]).[30]

#
# 3
Heck Reaction
The intramolecular Heck reaction is considered to be one of the most useful methods for the construction of medium-sized heterocycles due to its functional group tolerance and high stereoselectivity. In 2009, the Majumdar group developed an interesting and simple procedure for the synthesis of various annulated azocines from unactivated allylic substrates using a combination of two reactions, the aza-Claisen rearrangement and a palladium-catalyzed intramolecular Heck reaction. Thus, an appropriate substrate 59, prepared from N-allylanilines 58 by aza-Claisen rearrangement, tosylation, and alkylation with 2-bromobenzyl bromides, was subjected to the intramolecular Heck reaction to give exo-Heck cyclized products, dibenzo[b,f]azocines 60, in 72–79% yields (Scheme [18]).[31]

This strategy was successfully used for the synthesis of dibenzo[b,f]azocinones.[32] The precursors 61 for the Heck reaction were obtained from N-allyl-substituted anilines by aza-Claisen rearrangement, tosylation, and amidation with 2-iodobenzoyl chloride. The products of the Heck reaction depended on the conditions used. It was shown that Jeffrey’s two-phase protocol[32] led to endocyclic product 63 whereas phosphine-assisted standard conditions yielded exocyclic products 62 (Scheme [19]).

The same combined aza-Claisen rearrangement and intramolecular Heck reaction was successfully applied to the synthesis of coumarin- or quinolone-annulated azocines 65 [33] and pyrimidoazocines 67.[34] The precursors 64 for the Heck reaction were synthesized using aza-Claisen rearrangement of N-allylcoumarins or N-allylquinolones followed by alkylation with benzyl bromides. The intramolecular Heck reaction afforded azocine 65 in 75–79% yields (Scheme [20]).

In the case of pyrimido[5,4-b]azocines 67, the substrates 66 for the Heck reaction were prepared from 1,3-dialkyl-5-bromouracils by reaction with allylamine, subsequent aza-Claisen rearrangement, tosylation, and alkylation with 2-bromobenzyl bromide. Pyrimidoazocines 67 were obtained in high yields (Scheme [21]).


The Majumdar group achieved another efficient and straightforward method for the construction of dibenzoazocinones 72 and coumarin- and quinolone-annulated azocinones 73 via Pd-mediated reductive Mizoroki–Heck reaction.[35] The starting N-methyl or N-ethyl o-substituted amines 68 and 71 reacted with 2-iodophenylacetyl chloride giving amides 69 and 72, which were subjected to Heck reaction producing the 8-exocyclized products 70 and 73 in 42–60% yields (Scheme [22]).



Kim and co-workers obtained tetracyclic azocine systems 77 from indole derivatives 76 by applying the intramolecular palladium-catalyzed Heck reaction.[36] The required starting materials were synthesized by the reaction of Baylis–Hillman acetates 74 and indoles 75 (Scheme [23]).


Application of this protocol to compounds 79, 82, and 85, derived from the reaction of Baylis–Hillman acetate 74 with isatins 78, benzimidazoles 81, and carbazole 84, respectively, resulted in the formation of tetra(penta)cyclic azocines, benzo[4,5]azocino[3,2,1-hi]indoles 80 (Scheme [24]), benzo[e]imidazo[4,5,1-kl][1]benzoazocines 83 (Scheme [25]), and benzo[4,5]azocino[1,2,3-jk]carbazole 86 (Scheme [26]).[37]
Martin and co-workers synthesized azocino[3,4,5-cd]indoles 88 + 89 and 91 by Pd-catalyzed microwave-assisted Heck reaction from allylamine derivative 87 or enamine 90 (Scheme [27]).[38]
The unusual Heck product azocino[4,3-b]indole 93, resulting from an apparent 7-endo-cyclization with inversion of the ethylidene configuration, was obtained from cyclohepta[b]indole 92 in 43% yield (Scheme [28]).[39]
A readily separable mixture (1.3:1.0) of bridged benzoazocines 95 and 96 was formed through an intramolecular microwave-assisted Heck cyclization from azepine derivative 94 (Scheme [29]).[40]
Dibenzo[b,f]azocine 98 was produced via microwave-assisted Heck coupling from bifunctional precursor 97 prepared by vinylation of bromobenzaldehyde and subsequent reductive imine condensation with the relevant 2-bromoaniline.[41] Dibenzo[b,f]azocine 98 was also synthesized by a Suzuki–Heck cascade and also by a one-pot preparation (Scheme [30]).[41]



Starting from propargylamide 99, a novel protocol for the tandem Heck–Suzuki reaction was used for the construction of the benzoazocines 100 and 101 (Scheme [31]).[42]

# 4
Cycloaddition
In 2009, Rovis and co-workers developed the first enantioselective rhodium-catalyzed [4+2+2] cycloaddition of terminal alkynes 102 and dienyl isocyanates 103 leading to the formation of bicyclic azocines 104 and 105.[43] Pyrrolo[1,2-a]azocines 104 and 105 were obtained in good to high yields and excellent enantioselectivity (Scheme [32]). The geometry of the diene moiety had a significant effect on the selectivity of the products and used pure (E)-diene was used as the starting substrate.

A new and simple synthesis for azocine derivatives 108 by [6+2] cycloaddition reaction was suggested by Saito and co-workers.[44] Electron-deficient allenes 107 reacted with 2-vinylazetidine 106 giving azocines 108 in moderated yields (Scheme [33]).

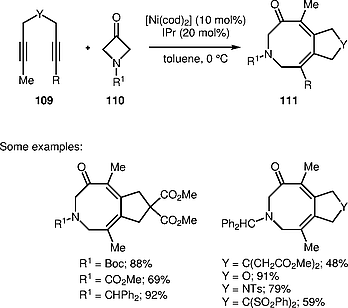
Louie and co-workers demonstrated in 2012 that azetidin-3-ones 110 under the action of diynes 109 underwent a Ni/IPr-catalyzed cycloaddition reaction leading to dihydroazocinones 111 (Scheme [34]).[45] The method involves a Csp2–Csp3 bond-cleavage step that proceeds at low temperatures.
In 2013, Louie and co-workers reported the Ni/P(p-Tol)3-catalyzed cycloaddition of 1,3-dienes 112 and azetidin-3-ones 113 yielding 1,4,7,8-tetrahydroazocin-2(3H)-ones 114 (Scheme [35]).[46]


Bower and co-workers reported a direct approach to substituted azocinediones 116 by a Rh-catalyzed cycloaddition–fragmentation process.[47] Exposure of N-cyclopropylacrylamides 115 to phosphine-ligated cationic Rh(I) catalyst systems under a CO atmosphere led to the formation of rhodacyclopentanone intermediates. The subsequent insertion of the alkene fragment into the intermediates was followed by fragmentation to give azocinediones 116 (Scheme [36]). The overall process is considered to be equivalent to a [7+1]-cycloaddition–tautomerization sequence.
# 5
Ring-Closing Metathesis (RCM)
Another powerful method for the construction of medium-sized nitrogen-containing systems that has received considerable attention in recent years is ring-closing metathesis.
Li and co-workers reported a five-step sequence for the construction of dibenzo[b,f]azocinone 119 where the key step was ruthenium-mediated ring-closing metathesis.[48] Starting from methyl 4-amino-3-iodobenzoate (117) the synthesis involved Stille coupling with tributyl(vinyl)stannane, followed by acylation with 2-vinylbenzoyl chloride, Boc protection, and RCM. Using Grubbs II catalyst, RCM of compound 118 resulted in dibenzo[b,f]azocinone 119 (Scheme [37]).

Starting from styryldiazoacetate 120, azocine 122 was obtained by a sequence of two reactions: N–H insertion and RCM.[49] It was also possible to combine the carbenoid N–H insertion and RCM reactions in a one-pot procedure for the synthesis of methyl 1,2,5,6,7,8-hexahydroazocine-2-carboxylate 122 (Scheme [38]).

Carbamate 124, obtained through condensation of aminobenzaldehyde 123 with methylamine and subsequent in situ reaction with benzyl chloroformate and then allylzinc bromide, underwent facile RCM in the presence of Grubbs II catalyst to give benzo[b]azocine 125 in 81% yield (Scheme [39]).[38]
The ring-closing metathesis approach was utilized to prepare novel 1,7-annulated azocino[3,2,1-hi]indole derivatives 129 starting from indoles 126 (Scheme [40]).[50] [51] Formylation of indole 126 followed by allylation of the N-atom and condensation with nitromethane led to 1-allyl-7-(2-nitrovinyl)indole 127. Reaction of 1-allyl-7-(2-nitrovinyl)indole 127 with allylmagnesium bromide gave 1-allyl-7-[1-(nitromethyl)but-3-enyl]indole 128 that underwent ring-closing metathesis to give azocinoindoles 129 in moderate yields.


Based on combined the aza-Claisen rearrangement and ring-closing metathesis, the Majumdar group obtained pyrimido[5,4-b]azocine derivatives 135 in excellent yields (Scheme [41]).[52] The starting 5-bromouracil derivatives 130 reacted with allylamine to give 5-allyluracils 131, subsequent catalyzed aza-Claisen rearrangement and tosylation led to tosyl derivatives 133. Reaction of 133 with homoallyl bromide provided the required precursor 134 for RCM using the Grubbs first-generation catalyst (Grubbs I) to give 135.

Using the same concept, the Majumdar group prepared RCM precursors 137 and 140. Starting from aminonaphthalenes 136 and 139, reaction with allylamine, subsequent aza-Claisen rearrangement, tosylation, and alkylation with homoallyl bromide gave 137 and 140. Under the RCM conditions, naphthalene derivatives 137 and 140 gave cyclized products, naphtho[1,2-b]azocines 138 and naphtho[2,1-b]azocines 141, respectively, in good yields (Scheme [42]).[53]

Lindsley and co-worker developed a novel six-step approach for the rapid and enantioselective synthesis of pyrrolo[1,2-a]azocines 147 and 152.[54] Commercially available aldehyde 142 was converted into (R)-N-sulfinylaldimine 143, followed by indium-mediated allylation yielding 144. Subsequent alkenylation of 144 with 5-bromopent-1-ene provides 145 which underwent RCM reaction with Grubbs II catalyst to deliver 146 in 70% yield for two steps. Hydrogenation, followed by a one-pot deprotection/acetal hydrolysis/reductive amination sequence produced decahydropyrrolo[1,2-a]azocine 147 in 87% yield for two steps and with more than 98% ee (Scheme [43]).

Pyrrolo[1,2-a]azocinone 152 was synthesized from commercial aldehyde 148, which was converted into (S)-N-sulfinylaldimine 149. Indium-mediated allylation of 149 to give 150 and subsequent deprotection gave a primary amine that cyclized to give (S)-5-allylpyrrolidin-2-one 151. The alkenylation of lactone 151 with 5-bromopent-1-ene, followed by RCM with Grubbs II catalyst afforded 1,5,6,7,10,10a-hexahydropyrrolo[1,2-a]azocin-3(2H)-one 152 in 73% yields for two steps (Scheme [44]).

In 2013, using a modified strategy Lindsley and co-workers developed a rapid route to access pyrrolo[1,2-a]-, pyrido[1,2-a]-, and azepino[1,2-a]azocines 153–155 (Scheme [45]).[55]

Benedetti, Penoni, and co-workers obtained benzo[c]azocine derivative 158 in 69% yield from enyne 157, from 156 using a Sonogashira reaction (Scheme [46]).[56]

Hexahydroazocine 161 was formed by microwave-assisted RCM of an α-allyl-α-phenyl-α-amino acid 160 obtained in two steps from α-imino ester 159 (Scheme [47]).[57]
Dash and co-workers constructed imidazo[1,5-a]azocine 163 from commercially available hydantoin 162 via a four-step procedure involving selective N-allylation and C5-alkylation and with the key step being RCM (Scheme [48]).[58]
Moss reported the efficient synthesis of a range of heterocycle-fused azocine derivatives 165–167 employing a directed metalation/ruthenium-catalyzed RCM approach.[59] The RCM precursors 164 were synthesized from carboxylic acids or 2-chloro-4-iodopyridine in three to four steps (Scheme [49]).

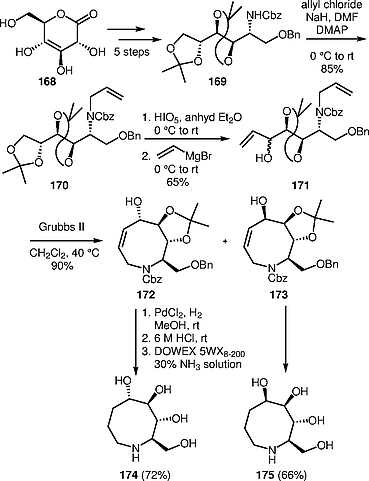
Rao and co-workers developed a common strategy for the construction of polyhydroxy azocine derivatives, including a novel example, from d-1,5-gluconolactone 168, using an RCM protocol as the key step.[60] d-1,5-Gluconolactone 168 was converted into compound 169 by a five-step procedure. Compound 169 was subjected to allylation with allyl chloride producing N-allyl derivative 170; subsequent deprotection, oxidative cleavage, and reaction with vinylmagnesium bromide gave 171. RCM of compound 171 afforded the cyclized products 172 and 173. The final deprotection and hydrogenation of azocines 172 and 173 provided polyhydroxy azocine derivatives 174 and 175 in 72% and 66% yields, respectively (Scheme [50]).


Bertozzi and Sletten synthesized a novel strained azacyclooctyne 181, which represents a new class of heterocyclic substrates for Cu-free click chemistry.[61] The synthesis involved nine steps beginning from 6-bromoglucopyranoside 176. First, compound 176 was transformed into acyclic diene 177 via zinc reduction/reductive amination reaction followed by amide formation with methyl succinyl chloride. The eight-membered ring was constructed by RCM giving azocine 178, which was converted into ketone 179 by oxidation and subsequent hydrogenation. The condensation of 179 with semicarbazide and then oxidation with selenium dioxide led to selenadiazole 180. Subsequent thermal decomposition followed by saponification of the ester produced azocine 181 (Scheme [51]).

In 2011, Danheiser and co-workers showed that the combination of ynamide-based benzannulation with RCM provides an expeditious strategy for the assembly of benzofused nitrogen heterocycles including azocines 187–189.[62] The precursors 184–186 for RCM were obtained via benzannulation from cyclobutenones with ynamide 183, prepared by reaction of carbamate 182 with 5-bromopent-1-en-4-yne. RCM occurred in the presence of the Grubbs II catalyst in dichloromethane (Scheme [52]).
The synthetic use of this strategy is illustrated in its application in a concise enantioselective route to the benzoazocine core 190 of the antitumor agents (+)-FR900482 and (+)-FR66979. The synthesis of the benzoazocine core 191 and the completion of the formal total synthesis of FR900482 and Fr66979 are shown in Scheme [53].
In 2013, Danheiser and co-workers employed the ‘second generation’ of benzannulation/RCM strategy, in which α-diazo ketones 192 were employed as vinylketene substrates instead of cyclobutenones.[63] Using this method hydroxy-substituted naphtho[2,3-b]azocine 193 was obtained in good yield (Scheme [54]).
In their investigations to develop concise total syntheses of some indole alkaloids possessing the azocine ring, Bennasar and co-workers successfully applied the combination of RCM and vinyl halide Heck cyclization for the construction of the azocinoindole moiety. The first unsuccessful attempt to work out the total synthesis of (±)-apparicine led to unexpected tetracycle 198.[64] The required RCM substrates 195a,b, prepared from 2-vinylindole-3-carbaldehyde 194 by reductive amination followed by N-acylation or sulfonylation, were subjected to RCM giving azocinoindoles 196,b in acceptable yields. The removal of the Boc group in azocinoindole 196a, subsequent alkylation with (Z)-2-iodobut-2-enyl tosylate and Heck cyclization yielded compound 198 possessing a bridged azocine ring (Scheme [55]).
Changing the cyclization site from 5,6-position to 4,5-position by using as a RCM precursor 2-allyl-3-[(allylamino)methyl]indole 199 and introducing an additional isomerization step before the Heck cyclization, they succeeded in accomplishing the first total synthesis of (±)-apparicine (Scheme [56]).[64]
The combination RCM/Heck cyclization successfully was utilized for the synthesis of the upper-half of vinorelbine.[65] Reductive amination of aldehyde 200 followed by Boc-protection of the aliphatic nitrogen gave carbamate 201, which smoothly underwent RCM in the presence of the Grubbs second-generation catalyst to give azocinoindole 202. The sequence of N-Boc deprotection, alkylation with allylic bromide, N-indole-deprotection, and Heck cyclization gave the upper-half of vinorelbine 205 (Scheme [57]).
RCM for the building of eight-membered nitrogen-containing cycle has also found application in the completion of the total synthesis of (–)-nakadomarin A, which shows interesting cytotoxic and antibacterial activity. A concise diastereoselective total synthesis was completed in 21 steps from d-pyroglutamic acid, wherein one of the key steps was the construction of the azocine ring via RCM (Scheme [58]).[66]
# 6
Cyclization
6.1Metal-Catalyzed Cyclization
Chowdhury and co-workers described an elegant method for the synthesis of benzo[c][1,2,3]triazolo[1,5-a]azocines 208 via palladium/copper-catalyzed heterocyclization.[67] The starting ortho-iodo azides 206 were prepared from the corresponding alcohols by mesylation and subsequent azidation. ortho-Iodo azides 206 underwent palladium/copper-catalyzed azide–alkyne cycloaddition with various terminal acetylenes 207 followed by arylation of the triazole to give azocine derivatives 208. Employing 1,3-diethynylbenzene (209) as a reactant, led to bis-heteroannulation giving azocine 210 in moderate yield (Scheme [59]). It is worth noting that the protocol included the formation of one C–C and two C–N bonds in a one-pot reaction.
Benzo[5,6]azocino[3,4-b]indoles 215 were obtained in four steps from indole 211 through an intramolecular direct arylation reaction as the key step.[68] Indole 211 was first treated with amine 212 to give amide 213; N-Boc-protection or N-methylation gave compounds 214. The cyclization of 214 mediated by Pd(0) delivered benzo[5,6]azocino[3,4-b]indoles 215 (Scheme [60]).




Pyrroloazocine 219 and azocinoindoles 217 were obtained via a palladium-catalyzed norbornene-mediated tandem process involving the intramolecular ortho-alkylation of an aromatic C–H bond followed by intramolecular direct arylation reaction from compounds 216 and 218, respectively (Scheme [61]).[69]

The Van der Eycken group, in 2009, developed a short and selective approach towards azocino[5,4-b]indoles 221 using a microwave-assisted Hg(OTf)2-catalyzed intramolecular carbocyclization of amides 220 prepared from corresponding tryptamines and 3-substituted prop-2-ynoic acids (Scheme [62]).[70]
In 2011, the Van der Eycken group elaborated a novel procedure for the construction of the interesting azocino[cd]indoles 224 via a Pd-catalyzed intramolecular acetylene hydroarylation.[71] The required for the cyclization, substrates 223 were synthesized by DCC-mediated amidation of suitable 4-bromotryptamines 222 and various propynoic acid derivatives. Microwave-assisted Pd-catalyzed cyclization of indoles 223 proceeded smoothly leading to regio- and stereoselective azocino[4,5,6-cd]indole derivatives 224 (Scheme [63]).
The Van der Eycken group have also reported the synthesis of azocinoindoles via an efficient gold-catalyzed post-Ugi intramolecular hydroarylation.[72] [73] Ugi-adducts 225 and 227 underwent 8-endo-dig cyclization leading to azocino[5,4,3-cd]indoles 226 and azocino[5,4-b]indoles 228, respectively. The merits of this method are good to excellent yields, a wide range of functional groups introduced during the Ugi reaction, and selectivity for 8-endo-dig cyclization (Scheme [64]).








Using a cationic gold-catalyzed intramolecular hydroarylation reaction of β-lactam-tethered allenyl indoles 229, Alcaide, Almendos, and co-workers obtained tetrahydroazeto[1′,2′;1,2]azocino[3,4-b]indoles 230 as single isomers in good yields (Scheme [65]).[74] The formation of azocines 230 was rationalized through an 8-endo carbocyclization of the indole group towards the terminal allene carbon. The gold-catalyzed cyclization allowed the regioselective formation of fused β-lactams without harming the sensitive four-membered heterocycle.

Pentacyclic azocine derivative 232 was generated in good yield via a novel gold-catalyzed cascade cyclization from N-[(2-azidophenyl)ethynyl]benzamides 231 (Scheme [66]).[75]
Echavarren and co-workers reported the synthesis of the azocino[5,4-b]indole core skeleton of the lundurines by gold-catalyzed 8-endo-dig cyclization of an alkynylindole.[76] The AuCl3-catalyzed cyclization of 2-[2-(2-ethynyl-5-oxopyrrolidino)ethyl]indole 233, prepared in seven steps from 2-(1H-indol-3-yl)acetate, afforded azocinoindole 234 in 55% isolated yield (Scheme [67]); the feasibility of using other gold complexes was considered.


# 6.2
Radical Cyclization
The Majumdar group developed a new efficient method for the synthesis of pyrimidoazocine derivatives 238 via the first example of an 8-endo-trig thiophenol-mediated radical cyclization.[77] The radical precursors 237 were prepared from pyrimidines 235, products of the aza-Claisen rearrangement of N-allyl-substituted pyrimidines, by tosylation and the subsequent reaction of intermediates 236 with propargyl bromide. The alkenyl radicals were generated from thiophenol initiated by benzoyl peroxide. The pyrimido[5,4-b]azocines 237 were obtained in excellent yields (Scheme [68]).

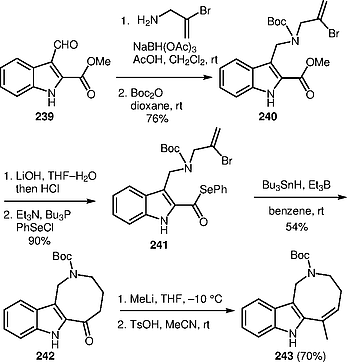
On developing the total synthesis of (±)-apparacine, Bennasar and co-workers prepared azocino[4,3-b]indole 242 via radical cyclization.[64] The synthesis of compounds 242 began with the preparation of selenoester 241 as the radical precursor. Selenoester 241 was prepared by reductive amination of aldehyde 239, followed by Boc protection of the resulting secondary amine, and phenylselenenation of the corresponding carboxylic acid. The treatment of selenoester 241 with Bu3SnH as the radical mediate and Et3B as the initiator led to the formation of azocinoindole 242. The reaction of 242 with methyllithium followed by dehydration of the intermediate tertiary alcohol provided azocinoindole 243 (Scheme [69]), which successfully used in the synthesis of (±)-apparacine (see Scheme [55]).
In their investigations, Li and co-workers generated azocine derivatives 245 and 247 starting from diesters 244 and 246 by a sequence of reactions in which the azocine-formation step was radical cyclization.[78] In the case of azocine 245 it was a Mn(III)-mediated oxidative radical process, whereas the azocine system 247 was obtained by a reductive radical process (Scheme [70]).

Diaba, Bonjoch, and co-worker obtained morphan compounds 249 via the first intramolecular atom transfer radical process between trichloroacetamide and enol acetate used as a radical acceptor.[79] The reaction was promoted by Grubbs II catalyst, thus expending the scope of these catalysts beyond the metathesis reaction (Scheme [71]).
This reaction enabled the construction of the tricyclic skeleton of the immunosuppressant FR901483. The required proradical trichloroacetamide 251 was synthesized in five steps starting from azaspirodecane 250. The treatment of ketone 251 with isopropenyl acetate gave a regioisomeric mixture of enol acetate 252 in a 1.8:1 ratio, the unseparated mixture was treated with Grubbs II catalyst affording the diazatricyclic derivative 253, its epimer 254, and unexpected mixture of enones 255. The reaction of compound 253 with zinc led to dechlorinated derivative 256, possessing the tricyclic skeleton of immunosuppressant FR901483, in 58% yield (Scheme [72]).[79]


Li and co-workers used a route based on the iodine-atom-transfer radical 8-endo cyclization to synthesize a number of azocine derivatives.[80] Thus, N-acyloxazolidinones 257 underwent 8-endo cyclization promoted by BF3·OEt2/H2O leading to the formation of oxazoloazocine 258 in high yields with excellent regio- and stereoselectivity (Scheme [73]). It is interesting to note that the product configuration was changed from 3,8-trans to 3,8-cis.
Li and co-workers also showed that in the presence of Mg(ClO4)2 and a bis(oxazoline) ligand, N-ethoxycarbonyl-substituted 2-iodo-N-(pent-4-enyl)alkanamides 259 underwent 8-endo cyclization giving only 3,5-trans-substituted azocan-2-ones 260 in excellent yields (Scheme [74]).


Similarly, the BF3·OEt2/H2O promote reaction of N-(2-allylaryl)-N-(ethoxycarbonyl)-2-iodoalkanamides 261 afforded benzo[b]azocines 262 with cis-3,5-configuration in high yields (Scheme [75]).

# 6.3
Friedel–Crafts Cyclization
Azocino[3,4-b]indol-1-ones 264 were obtained via acid-catalyzed intramolecular Friedel–Crafts cyclization of 1-methyl-N-(4-oxobutyl)indole-2-carboxamides 263 in low yields (Scheme [76]).[81]

Pandey and co-workers developed a general route for the synthesis of azocino[5,4-b]indoles 269 starting from allyl bromide 265 which prepared from Morita–Baylis–Hillman adducts.[82] The reaction of tryptamine with allyl bromide 265 gives 266, protection of the nitrogen atoms in 266 gives 267, and saponification of the ester group in 267 gave indoles 268 which underwent Friedel–Crafts intramolecular cyclization affording azocino[5,4-b]indoles 269 in good yields (Scheme [77]).

Kim and Seo used the Friedel–Crafts reaction as the key step for the construction of azocine ring in the tetracyclic compound 274.[83] Aminoacrylate 271, prepared from 3,4-dimethoxyphenethylamine via amidation reaction with 2-iodobenzoic acid and subsequent Michael addition of ethyl propynoate, was subjected to the Heck reaction giving isoindole 272. The hydrogenation of isoindole 272 followed by hydrolysis provide derivative 273 which by the action of polyphosphoric acid was converted into tetracyclic compound 274 (Scheme [78]).

The same sequence of reactions was used in the case of the synthesis of an azocine alkaloid, magallanesine (Scheme [79]).[83]

# 6.4
Other Examples of Cyclization
In a new synthetic route for the synthesis of the dasycarpidone skeleton and for the total synthesis of (±)-uleine, Patir and Uludag used acid-catalyzed intramolecular cyclization to construct the azocino[4,3-b]indole core.[84] Reduction of ketoamide 275 with borane–dimethyl sulfide complex and the subsequent acidification of the resulted alcohol 276 with TFA led to 277, which was treated in situ with DDQ furnishing the desired tetracyclic compounds 278 in good yield. Four further steps were required to complete the total synthesis of (±)-uleine from azocinoindole 278 (Scheme [80]).

Azocinoindoles 280 and 283 were obtained by Hamada and co-workers via a novel acid-promoted skeletal rearrangement of 2- or 3-alkylideneindolenium cations generated from compounds 279 and 282.[85] [86] A reaction cascade leading to the azocine system involved intramolecular ipso-Friedel–Crafts alkylation of phenols, rearomatization of the spirocyclohexadienone unit, and iso-Pictet–Spengler reaction. In addition to the targeted azocinoindoles, pyrrolidine derivatives 281 and 284 were isolated as the major byproducts (Scheme [81]).

Azocino[4,3-b]indoles 287 were synthesized by the oxidative cyclization of [3-(3-oxopiperidin-2-yl)indol-2-yl]malonates 286 obtained in four steps from dimethyl or di-tert-butyl malonates 285.[87] Furthermore, these azocino[4,3-b]indoles were successfully used for the total synthesis of (±)- and (–)-actinophyllic acid (Scheme [82]).

Reitz and co-workers have prepared benzo[d]azocines 294 and 295 which are formally analogues of Phe-Ala or Ala-Phe dipeptides joined together on their side chains.[88] The synthesis began with the conversion of the N-Boc-protected 2′-iodo-l-phenylalanine 288 into N-Fmoc derivative 289. Negishi coupling of 289 with either Boc-(β-I)-d-Ala-OMe or Boc-(β-I)-l-Ala-OMe gave dipeptides 290 and 291, respectively. Removals of Boc- and benzyl ester groups of bis-amino acids 290 and 291 and amide formation and cyclization with 1-hydroxy-7-azabenzotriazole (HOAt) and O-(7-azabenzotriazole-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) provided azocines 292 and 293. Fmoc-deprotection followed by simultaneous amidolysis of the ester with methylamine and acetylation led to dipeptides 294 and 295 (Scheme [83]).


Hexahydrochromeno[3,4-c]azocine 297 was obtained from chromene 296 by cyclization which occurred during Duff formylation, but the yield of the cyclic product was poor at only 9% (Scheme [84]).[89]
#
# 7
Microwave- and Photo-Assisted Reactions
Alcaide, Almendos, and co-workers developed a method for the synthesis of structurally novel bicyclic azocine-fused β-lactams 299 and 300 in the absence of any metal catalyst.[90] This was the first example of metal-free preparation of eight-membered rings by the thermolysis of nonconjugated azetidin-2-one-tethered bis(allenes) 298 on application of microwave irradiation. Azocines 299 and 300 were isolated as single regio- and diastereoisomers (Scheme [85]).

The Van der Eycken group elaborated a new approach towards the construction of 5,6,7,8-tetrahydrodibenzo[c,e]azocines 303 via a microwave-assisted copper-catalyzed intramolecular A3-coupling reaction.[91] Formed in situ by Boc deprotection of biaryl compounds 301, biaryl derivatives 302 with both amino and aldehyde groups reacted with the suitable alkynes in the presence of CuBr under focused microwave irradiation thus forming dibenzo[c,e]azocines 303 in good to excellent yields (Scheme [86]).

Yudin and Cheung found that N-vinyl-β-lactams 304 underwent microwave-assisted ring-expansion, resulting from [3,3]-sigmatropic rearrangement between two strategically placed alkene moieties on the β-lactam, giving azocines 305 in yields of 8–86% (Scheme [87]).[92] [93]

On irradiation of 4-diazo-4H-imidazole 306 in hexafluorobenzene gave the unusual imidazo[3,4-a]azocine 307 in 51% yield (Scheme [88]).[94]

Kutateladze and co-workers synthesized epoxybenzoazocines 309 and 311 from aniline derivatives 308 and 310, respectively, by photo-generation of azaxylylenes and their subsequent intramolecular [4+4] cycloaddition with a furan-containing pendant tethered either via the aniline nitrogen or through the carbonyl-group-containing fragment (Scheme [89]).[95]

# 8
Other Methods
8.1Cascade and Tandem Reactions
Nakamura and co-workers elaborated an efficient synthesis of azocine derivatives 313 from O-propargylic oximes 312 in good to excellent yields by the means of a Rh-catalyzed 2,3-rearrangement/heterocyclization cascade sequence.[96] It is noteworthy that the chirality of the substrate was maintained throughout the cascade process to afford optically active azocines 313d (Scheme [90]).

She, Xie, and co-workers accomplished a gold-catalyzed 1,2-acyloxy migration/intramolecular [3+2]-cycloaddition cascade reaction for the construction of unsaturated azocines 315 starting from enynyl esters 314 (Scheme [91]).[97]

In 2016, She, Xie, and co-workers subsequently expanded this gold-catalyzed 1,2-acyloxy migration/intramolecular [3+2]-cycloaddition cascade reaction to the synthesis of benzo[d]azocines 317 from 1,9-enynyl esters 316. The reaction proceeded under mild conditions leading to benzoazocines 317 in good to excellent yields of 55–82% (Scheme [92]).[98]

Kumar and co-workers synthesized benzo[b]azocines 318 through an unprecedented one-pot, triflic acid mediated, tandem Michael addition–Fries rearrangement of sorbylanilides 319.[99] The reaction is proposed to proceed via a δ-lactam intermediate, earlier considered unreactive for the Fries rearrangement (Scheme [93]).
In their research concerning the total synthesis of (±)-actinophyllic acid, Martin and co-workers constructed the tetracyclic core of the natural compound in a single chemical operation via a novel Lewis acid catalyzed cascade of reactions involving stabilized carbocations and π-nucleophiles.[100] The treatment of a mixture of electrophile precursor indole 320 and π-nucleophiles 321 with TMSOTf in the presence of 2,6-di-tert-butylpyridine, followed by addition of NaOMe in MeOH at –78 °C gave tetracyclic systems 322 in 53–80% yields (Scheme [94]).


# 8.2
Aldol Condensation
In their work on the total syntheses of (–)-FR901483 and (+)-8-epi-FR901483, Huang and co-workers successfully used the aldol condensation for the construction of the azocine ring.[101] [102] Starting from the known chiron (R)-1-allyl-3-benzyloxypiperidine-2,5-dione, piperidin-3-ol 324 was obtained in four steps. The oxidation of 323 gave a ketone that underwent an intramolecular aldol ring-closure reaction forming azocine derivative 324 (Scheme [95]).

Uludag and co-workers ring-closed 1-oxo-1,2,3,4-tetrahydrocarbazole 325 by a NaH-promoted intramolecular aldol condensation to give the azocino[4,3-b]indole system 326 (Scheme [96]).[103]
# 8.3
Thermolysis
Thermolysis of hydrazine 327 in m-xylene under reflux led to the impressive cyclopropa[3,4]azocino[1,2-a]benzimidazole 328 in a poor yield of 11% with the two other products 329 and 330 in higher yields of 22% and 33% (Scheme [97]).[104]


Thermolysis of the 12-membered ring aza-enediyne 331 in benzene in the presence of catalytic amounts of p-toluenesulfonic acid produced the addition-dehydration product, naphtho[2,3-b]azocine 332 in 18% yield, however, the major product of this reaction was N-tosyl lactam 333 in 28% yield (Scheme [98]).[105]

# 8.4
Ring Opening
Tetracyclic azocine derivative 336 possessing a paullone-like structural framework was obtained in a single step from a novel 2,2′-spirobi[indolin]-3-one 335 prepared by Cu-mediated intramolecular cascade reaction of cyclopenta[b]indole 334.[106] Compound 335 when treated with methanolic KOH underwent demesylation followed by ring opening and subsequent aromatization to give 336 in 90% yield (Scheme [99]).

Yavari and Seyfi found that furo[2′,3′:2,3]cyclopenta[1,2-b]pyrroles 338, obtained by Wittig reaction from oxoindeno[1,2-b]pyrroles 337 and DMAD, underwent Et3N-mediated ring opening thus affording tetrahydrobenzo[c]furo[3,2-e]azocines 339 in good yields (Scheme [100]).[107]

# 8.5
Other Methods
Waghmode and co-workers synthesized epoxy-bridged benzo[d]azocines 342 in good to excellent yields from 1-(bromomethyl)-3-(tosyloxy)chromane 340 via nucleophilic substitution with various benzylamine derivatives 341 (Scheme [101]).[108]

Jia and co-workers elaborated a highly enantioselective palladium/l-proline-catalyzed α-arylative desymmetrization of cyclohexanones 343 leading to a series of optically active morphan derivatives 344 with α-carbonyl tertiary stereocenters in good yields (Scheme [102]).[109]

Xu, Li, and co-workers have designed and synthesized novel neonicotinoid analogues 346–348 with an aza-bridged azocine fragment.[110] Azocine derivatives 346–348 were prepared by reaction of imidazole 345 with glutaraldehyde and a primary amine hydrochloride (aliphatic amines, phenylhydrazines, and anilines) (Scheme [103]).

Azocine derivative 350 was obtained in 16% yield via cationic aza-Cope rearrangement of aminoketal 349 (Scheme [104]).[111]
Yao, Wu, and co-workers developed a novel facile and efficient route for the synthesis of benzo[b]naphtha[2,3-d]azocinones 353 through a palladium-catalyzed reaction of 2-alkynylanilines 351 with 2-(2-bromobenzylidene)cyclobutanones 352.[112] During the reaction process, double carbometalation resulting in the formation of three new bonds was involved (Scheme [105]).
Boeckman and co-workers obtained azocine derivatives 355 and 357 using a one-pot, aza-Wittig/retro-aza-Claisen sequence from 2-vinylcyclobutanecarbaldehydes 354 and 356, respectively.[113] The rearrangement sequence proceeded under mild conditions affording azocines 355 and 357 in 75–92% yields (Scheme [106]).
Modification of a previously reported procedure[114] allowed Raffa and co-workers obtain the polycyclic system, 5,7:7,12-dimethanopyrazolo[3,4-b]pyrazolo[3′,4′:2,3]azepino[4,5-f]azocine 360.[115] Methylaminopyrazoles 358 and hexane-2,5-dione (359) reacted in refluxing 1,4-dioxane in the presence of p-toluenesulfonic acid thus leading to compounds 360 in 10–37% yields (Scheme [107]).
Systematically investigating the reactivity of the palladacycles obtained in their studies, Vicente, Saura-Llamas, and co-workers synthesized various azocine-containing systems. Thus, heating of complex 361 in the presence of TlOTf and 2,4-dimethylphenyl isocyanide gave azocine 363 through insertion of isocyanide and C–N coupling process (Scheme [108]).[116]
The treatment of eight-membered palladacycles 364 and palladacycles 366 with CO afforded benzo[d]azocine-2,4-(1H,3H)-diones 365 [117] or hexahydrobenzo[d]azocinones 367,[118] which resulted from the insertion of a molecule of CO into the Pd–C bond and subsequent C–N reductive coupling (Scheme [109]).
These methods were extended to the preparation of dibenzo[c,e]azocines 369/370 and 371 via insertion of CO and isocyanide, respectively (Scheme [110]).[119]
#
# 9
Conclusion
In recent years, many new pathways towards eight-membered azaheterocycles have been elaborated including domino approaches, MCRs, metal-catalyzed cyclizations, RCM, and ring-expansion strategies. These approaches provide environmentally friendly and step-economical access towards several annulated azocines with substantial biological activity and natural compounds. However, much work remains to be done to elaborate general synthetic strategy towards medium-sized nitrogen heterocycles including azocines.







#
#
-
References
- 1 Voskressensky LG. Kulikova LN. Borisova TN. Varlamov AV. Adv. Heterocycl. Chem. 2008; 96: 81
- 2 Rössle M. Christoffers J. Synlett 2006; 106
- 3 Behler F. Habecker F. Saak W. Klüner T. Christoffers J. Eur. J. Org. Chem. 2011; 4231
- 4 Penning M. Christoffers J. Eur. J. Org. Chem. 2014; 2140
- 5 Penning M. Aeissen E. Christoffers J. Synthesis 2015; 47: 1007
- 6 Varlamov AV. Borisova TN. Voskressensky LG. Soklakova TA. Kulikova LN. Chernyshov AA. Alexandrov GG. Tetrahedron Lett. 2002; 43: 6767
- 7 Voskressensky LG. Borisova TN. Ovcharov MV. Kulikova LN. Sorokina EA. Borisov RS. Varlamov AV. Chem. Heterocycl. Compd. 2008; 44: 1510
- 8 Voskressensky LG. Borisova TN. Ovcharov MV. Sorokina EA. Khrustalev VN. Varlamov AV. Russ. Chem. Bull. 2012; 61: 1603
- 9 Voskressensky LG. Kovaleva SA. Borisova TN. Listratova AV. Eresko AB. Tolkunov VS. Tolkunov SV. Varlamov AV. Tetrahedron 2010; 66: 9421
- 10 Voskressensky LG. Ovcharov MV. Borisova TN. Kulikova LN. Listratova AV. Borisov RS. Varlamov AV. Chem. Heterocycl. Compd. 2011; 47: 222
- 11 Voskressensky LG. Ovcharov MV. Borisova TN. Listratova AV. Kulikova LN. Sorokina EA. Gromov SP. Varlamov AV. Chem. Heterocycl. Compd. 2013; 49: 1180
- 12 Voskressensky LG. Kovaleva SA. Borisova TN. Eresko AB. Tolkunov VS. Tolkunov SV. Listratova AV. Candia M. Altomare C. Varlamov AV. Chem. Heterocycl. Compd. 2013; 49: 930
- 13 Voskressensky LG. Borisova TN. Chervyakova TM. Matveeva MD. Galaktionova DV. Tolkunov SV. Tolkunova VS. Eresko AB. Varlamov AV. Chem. Heterocycl. Compd. 2014; 50: 1338
- 14 Voskressensky LG. Borisova TN. Chervyakova TM. Titov AA. Kozlov AV. Sorokina EA. Samavati R. Varlamov AV. Chem. Heterocycl. Compd. 2014; 50: 658
- 15 Voskressensky LG. Titov AA. Kobzev MS. Samavati R. Borisov RS. Kulikova LN. Varlamov AV. Chem. Heterocycl. Compd. 2016; 52: 68
- 16 Voskressensky LG. Borisova TN. Kovaleva SA. Listratova AV. Kulikova LN. Khrustalev VN. Ovcharov MV. Varlamov AV. Russ. Chem. Bull. 2012; 61: 370
- 17 Voskressensky LG. Kovaleva SA. Borisova TN. Titov AA. Toze F. Listratova AV. Khrustalev VN. Varlamov AV. Chem. Heterocycl. Compd. 2015; 51: 17
- 18a Borisov RS. Voskressensky LG. Polyakov AI. Borisova TN. Varlamov AV. Synlett 2014; 25: 0955
- 18b Zubkov FI. Ershova JD. Orlova AA. Zaytsev VP. Nikitina EV. Peregudov AS. Gurbanov AV. Borisov RS. Khrustalev VN. Maharramov AM. Varlamov AV. Tetrahedron 2009; 65: 3789
- 19 Voskressensky LG. Listratova AV. Borisova TN. Kovaleva SA. Borisov RS. Varlamov AV. Tetrahedron 2008; 64: 10443
- 20 Voskressensky LG. Kulikova LN. Gozun SV. Khrustalev VN. Borisova TN. Listratova AV. Ovcharov MV. Varlamov AV. Tetrahedron Lett. 2011; 52: 4189
- 21 Voskressensky LG. Borisov RS. Kulikova LN. Kleimenov AV. Borisova TN. Varlamov AV. Chem. Heterocycl. Compd. 2012; 48: 680
- 22 Cho H. Iwama Y. Sugimoto K. Kwon E. Tokuyama H. Heterocycles 2009; 78: 1183
- 23 Cho H. Iwama Y. Sugimoto K. Mori S. Tokuyama H. J. Org. Chem. 2010; 75: 627
- 24 Cho H. Tetrahedron 2014; 70: 3527
- 25 Cho H. Iwama Y. Okano K. Tokuyama H. Synlett 2013; 24: 813
- 26 González-Gómez Á. Domónguez G. Pérez-Castells J. Tetrahedron 2009; 65: 3378
- 27 Zhang L. Chang L. Hu H. Wang H. Yao Z.-J. Wang S. Chem. Eur. J. 2014; 20: 2925
- 28 Harthong S. Bach R. Besnard C. Guénée L. Lacour J. Synthesis 2013; 45: 2070
- 29 Mohammadizadeh MR. Alborz M. Tetrahedron Lett. 2014; 55: 6808
- 30 Malkova AV. Polyanskii KB. Soldatenkov AT. Soldatova SA. Merkulova NL. Khrustalev VN. Russ. J. Org. Chem. 2016; 52: 219
- 31 Majumdar KC. Chattopadhyay B. Samanta S. Tetrahedron Lett. 2009; 50: 3178
- 32 Majumdar KC. Samanta S. Chattopadhyay B. Tetrahedron Lett. 2009; 50: 4866
- 33 Majumdar KC. Samanta S. Chattopadhyay B. Nandi RK. Synthesis 2010; 985
- 34 Majumdar KC. Mondal S. Ghosh D. Chattopadhyay B. Synthesis 2010; 1315
- 35 Majumdar KC. Ghosh T. Chakravorty S. Tetrahedron Lett. 2010; 51: 3372
- 36 Lee HS. Kim SH. Kim TH. Kim JN. Tetrahedron Lett. 2008; 49: 1773
- 37 Lee HS. Kim SH. Gowrisankar S. Kim JN. Tetrahedron 2008; 64: 7183
- 38 Sunderhaus JD. Dockendorff C. Martin SF. Tetrahedron 2009; 65: 6454
- 39 Bennasar M.-L. Zulaica E. Solé D. Alonso S. Tetrahedron 2012; 68: 4641
- 40 Sahn JJ. Martin SF. Tetrahedron Lett. 2011; 52: 6855
- 41 Jäger M. Görls H. Günther W. Schubert US. Chem. Eur. J. 2013; 19: 2150
- 42 Peshkov AA. Peshkov VA. Pereshivko OP. Van Hecke K. Kumar R. Van der Eycken EV. J. Org. Chem. 2015; 80: 6598
- 43 Yu RT. Friedman RK. Rovis T. J. Am. Chem. Soc. 2009; 131: 13250
- 44 Koya S. Yamanoi K. Yamasaki R. Azumaya I. Masu H. Saito S. Org. Lett. 2009; 11: 5438
- 45 Kumar P. Zhang K. Louie J. Angew. Chem. Int. Ed. 2012; 51: 8602
- 46 Thakur A. Facer ME. Louie J. Angew. Chem. Int. Ed. 2013; 52: 12161
- 47 Shaw MH. Croft RA. Whittingham WG. Bower JF. J. Am. Chem. Soc. 2015; 137: 8054
- 48 Hassam M. More SV. Li W.-S. Mendeleev Commun. 2014; 24: 159
- 49 Pavlyuk O. Teller H. McMills MC. Tetrahedron Lett. 2009; 50: 2716
- 50 Wood K. Black DSt. C. Namboothiri IN. N. Kumar N. Tetrahedron Lett. 2010; 51: 1606
- 51 Wood K. Black DSt. C. Kumar N. Tetrahedron 2011; 67: 4093
- 52 Majumdar KC. Mondal S. Ghosh D. Synthesis 2010; 1176
- 53 Majumdar KC. Samanta S. Chattopadhyay B. Nandi RK. Synthesis 2010; 863
- 54 Fadeyi OO. Senter TJ. Hahn KN. Lindsley CW. Chem. Eur. J. 2012; 18: 5826
- 55 Senter TJ. Schulte ML. Konkol LC. Wadzinski TE. Lindsley CW. Tetrahedron Lett. 2013; 54: 1645
- 56 Benedetti E. Lomazzi M. Tibiletti F. Goddard J.-P. Fensterbank L. Malacria M. Palmisano G. Penoni A. Synthesis 2012; 44: 3523
- 57 Curto JM. Kozlowski MC. J. Org. Chem. 2014; 79: 5359
- 58 Dhara K. Midya GC. Dash J. J. Org. Chem. 2012; 77: 8071
- 59 Moss TA. Tetrahedron Lett. 2013; 54: 4254
- 60 Jagadeesh Y. Ramakrishna K. Rao BV. Tetrahedron: Asymmetry 2012; 23: 697
- 61 Sletten EM. Bertozzi CR. Org. Lett. 2008; 10: 3097
- 62 Mak XY. Crombie AL. Danheiser RL. J. Org. Chem. 2011; 76: 1852
- 63 Willumstad TP. Haze O. Mak XY. Lam TY. Wang Y.-P. Danheiser RL. J. Org. Chem. 2013; 78: 11450
- 64 Bennasar M.-L. Zulaica E. Solé D. Roca T. García-Díaz D. Alonso S. J. Org. Chem. 2009; 74: 8359
- 65 Bennasar M.-L. Solé D. Zulaica E. Alonso S. Tetrahedron 2013; 69: 2534
- 66 Nilson MG. Funk RL. Org. Lett. 2010; 12: 4912
- 67 Brahma K. Achari B. Chowdhury C. Synthesis 2013; 45: 545
- 68 Putey A. Popowycz F. Do Q.-T. Bernard P. Talapatra SK. Kozielski F. Galmarini CM. Joseph B. J. Med. Chem. 2009; 52: 5916
- 69 Blaszykowski C. Aktoudianakis E. Alberico D. Bressy C. Hulcoop DG. Jafarpour F. Joushaghani A. Laleu B. Lautens M. J. Org. Chem. 2008; 73: 1888
- 70 Donets PA. Van Hecke K. Van Meervelt LE. Van der Eycken V. Org. Lett. 2009; 11: 3618
- 71 Peshkov VA. Van Hove S. Donets PA. Pereshivko OP. Van Hecke K. Van Meervelt L. Van der Eycken EV. Eur. J. Org. Chem. 2011; 1837
- 72 Kumar A. Li Z. Sharma SK. Parmar VS. Van der Eycken EV. Chem. Commun. 2013; 49: 6803
- 73 Modha SG. Vachhani DD. Jacobs J. Van Meervelt L. Van der Eycken EV. Chem. Commun. 2012; 48: 6550
- 74 Alcaide B. Almendros P. Cembellín S. del Campo TM. J. Org. Chem. 2015; 80: 4650
- 75 Tokimizu Y. Oishi S. Fujii N. Ohno H. Org. Lett. 2014; 16: 3138
- 76 Ferrer C. Escribano-Cuesta A. Echavarren AM. Tetrahedron 2009; 65: 9015
- 77 Majumdar KC. Mondal S. Ghosh D. Tetrahedron Lett. 2010; 51: 348
- 78 Zhang J. Wang Y.-Q. Wang X.-W. Li W.-DZ. J. Org. Chem. 2013; 78: 6154
- 79 Diaba F. Martínez-Laporta A. Bonjoch J. J. Org. Chem. 2014; 79: 9365
- 80 Fang X. Liu K. Li C. J. Am. Chem. Soc. 2010; 132: 2274
- 81 Cincinelli R. Dallavalle S. Merlini L. Nannei R. Scaglioni L. Tetrahedron 2009; 65: 3465
- 82 Pandey G. Kant R. Batra S. Tetrahedron Lett. 2015; 56: 930
- 83 Seo S.-Y. Kim G. Tetrahedron Lett. 2015; 56: 3835
- 84 Patir S. Uludag N. Tetrahedron 2009; 65: 115
- 85 Yokosaka T. Nemoto T. Hamada Y. Chem. Commun. 2012; 48: 5431
- 86 Yokosaka T. Kanehira T. Nakayama H. Nemoto T. Hamada Y. Tetrahedron 2014; 70: 2151
- 87 Martin CL. Overman LE. Rohde JM. J. Am. Chem. Soc. 2010; 132: 4894
- 88 Falcon BV. Creighton CJ. Parker MH. Reitz AB. Synth. Commun. 2008; 38: 411
- 89 Ranatunga S. Tang C.-HA. Kang CW. Kriss CL. Kloppenburg BJ. Hu C.-CA. Del Valle JR. J. Med. Chem. 2014; 57: 4289
- 90 Alcaide B. Almendros P. Aragoncillo C. Fernandez I. Gomez-Campillos G. J. Org. Chem. 2014; 79: 7075
- 91 Bariwal JB. Ermolat’ev DS. Glasnov TN. Van Hecke K. Mehta VP. Van Meervelt L. Kappe CO. Van der Eycken EV. Org. Lett. 2010; 12: 2774
- 92 Cheung LL. W. Yudin AK. Org. Lett. 2009; 11: 1281
- 93 Cheung LL. W. Yudin AK. Chem. Eur. J. 2010; 16: 4100
- 94 Smith MR. Blake AJ. Hayes CJ. Stevens MF. G. Moody CJ. J. Org. Chem. 2009; 74: 9372
- 95 Mukhina OA. Kumar NN. B. Cowge TM. Kutateladze AG. J. Org. Chem. 2014; 79: 10956
- 96 Nakamura I. Sato Y. Takeda K. Terada M. Chem. Eur. J. 2014; 20: 10214
- 97 Zhao C. Xie X. Duan S. Li H. Fang R. She X. Angew. Chem. Int. Ed. 2014; 53: 10789
- 98 Feng S. Wang Z. Zhang W. Xie X. She X. Chem. Asian J. 2016; 11: 2167
- 99 Anand A. Singh P. Mehra V. Amandeep, Kumar V. Mahajan MP. Tetrahedron Lett. 2012; 53: 2417
- 100 Granger BA. Jewett IT. Butler JD. Martin SF. Tetrahedron 2014; 70: 4094
- 101 Huo H.-H. Zhang H.-K. Xia X.-E. Huang P.-Q. Org. Lett. 2012; 14: 4834
- 102 Huo H.-H. Xia X.-E. Zhang H.-K. Huang P.-Q. J. Org. Chem. 2013; 78: 455
- 103 Uludag N. Yılmaz R. Asutay O. Colak N. Chem. Heterocycl. Compd. 2016; 52: 196
- 104 Hehir S. O’Donovan L. Carty MP. Aldabbagh F. Tetrahedron 2008; 64: 4196
- 105 Poloukhtine A. Rassadin V. Kuzmin A. Popik VV. J. Org. Chem. 2010; 75: 5953
- 106 Prasad B. Adepu R. Sharma AK. Pal M. Chem. Commun. 2015; 51: 1259
- 107 Yavari I. Seyfi S. Synlett 2012; 23: 1209
- 108 Patil VP. Ghosh A. Sonavane U. Joshi R. Sawant R. Jadhav S. Waghmode SB. Tetrahedron: Asymmetry 2014; 25: 489
- 109 Liu R.-R. Li B.-L. Lu J. Shen C. Gao J.-R. Jia Y.-X. J. Am. Chem. Soc. 2016; 138: 5198
- 110 Xu R. Xia R. Luo M. Xu X. Cheng J. Shao X. Li Z. J. Agric. Food Chem. 2014; 62: 381
- 111 Aron ZD. Ito T. May TL. Overman LE. Wang J. J. Org. Chem. 2013; 78: 9929
- 112 Gong X. Chen M. Yao L. Wu J. Chem. Asian. J. 2016; 11: 1613
- 113 Boeckman RK. Jr. Genung NE. Chen K. Ryder TR. Org. Lett. 2010; 12: 1628
- 114 Daidone G. Sprio V. Plescia S. Marino ML. Bombieri G. Bruno G. J. Chem. Soc., Perkin Trans. 2 1989; 137
- 115 Maggio B. Raffa D. Raimondi MV. Cusimano MG. Plescia F. Cascioferro S. Cancemi G. Lauricella M. Carlisi D. Daidone G. Eur. J. Med. Chem. 2014; 72: 1
- 116 Vicente J. Saura-Llamas I. Garcıa-López J.-A. Bautista D. Organometallics 2010; 29: 4320
- 117 Frutos-Pedreno R. Gonzalez-Herrero P. Vicente J. Organometallics 2012; 31: 3361
- 118 García-Lopez J.-A. Oliva-Madrid M.-J. Saura-Llamas I. Bautista D. Vicente J. Organometallics 2012; 31: 6351
- 119 Oliva-Madrid M.-J. García-Lopez J.-A. Saura-Llamas I. Bautista D. Vicente J. Organometallics 2014; 33: 6420
-
References
- 1 Voskressensky LG. Kulikova LN. Borisova TN. Varlamov AV. Adv. Heterocycl. Chem. 2008; 96: 81
- 2 Rössle M. Christoffers J. Synlett 2006; 106
- 3 Behler F. Habecker F. Saak W. Klüner T. Christoffers J. Eur. J. Org. Chem. 2011; 4231
- 4 Penning M. Christoffers J. Eur. J. Org. Chem. 2014; 2140
- 5 Penning M. Aeissen E. Christoffers J. Synthesis 2015; 47: 1007
- 6 Varlamov AV. Borisova TN. Voskressensky LG. Soklakova TA. Kulikova LN. Chernyshov AA. Alexandrov GG. Tetrahedron Lett. 2002; 43: 6767
- 7 Voskressensky LG. Borisova TN. Ovcharov MV. Kulikova LN. Sorokina EA. Borisov RS. Varlamov AV. Chem. Heterocycl. Compd. 2008; 44: 1510
- 8 Voskressensky LG. Borisova TN. Ovcharov MV. Sorokina EA. Khrustalev VN. Varlamov AV. Russ. Chem. Bull. 2012; 61: 1603
- 9 Voskressensky LG. Kovaleva SA. Borisova TN. Listratova AV. Eresko AB. Tolkunov VS. Tolkunov SV. Varlamov AV. Tetrahedron 2010; 66: 9421
- 10 Voskressensky LG. Ovcharov MV. Borisova TN. Kulikova LN. Listratova AV. Borisov RS. Varlamov AV. Chem. Heterocycl. Compd. 2011; 47: 222
- 11 Voskressensky LG. Ovcharov MV. Borisova TN. Listratova AV. Kulikova LN. Sorokina EA. Gromov SP. Varlamov AV. Chem. Heterocycl. Compd. 2013; 49: 1180
- 12 Voskressensky LG. Kovaleva SA. Borisova TN. Eresko AB. Tolkunov VS. Tolkunov SV. Listratova AV. Candia M. Altomare C. Varlamov AV. Chem. Heterocycl. Compd. 2013; 49: 930
- 13 Voskressensky LG. Borisova TN. Chervyakova TM. Matveeva MD. Galaktionova DV. Tolkunov SV. Tolkunova VS. Eresko AB. Varlamov AV. Chem. Heterocycl. Compd. 2014; 50: 1338
- 14 Voskressensky LG. Borisova TN. Chervyakova TM. Titov AA. Kozlov AV. Sorokina EA. Samavati R. Varlamov AV. Chem. Heterocycl. Compd. 2014; 50: 658
- 15 Voskressensky LG. Titov AA. Kobzev MS. Samavati R. Borisov RS. Kulikova LN. Varlamov AV. Chem. Heterocycl. Compd. 2016; 52: 68
- 16 Voskressensky LG. Borisova TN. Kovaleva SA. Listratova AV. Kulikova LN. Khrustalev VN. Ovcharov MV. Varlamov AV. Russ. Chem. Bull. 2012; 61: 370
- 17 Voskressensky LG. Kovaleva SA. Borisova TN. Titov AA. Toze F. Listratova AV. Khrustalev VN. Varlamov AV. Chem. Heterocycl. Compd. 2015; 51: 17
- 18a Borisov RS. Voskressensky LG. Polyakov AI. Borisova TN. Varlamov AV. Synlett 2014; 25: 0955
- 18b Zubkov FI. Ershova JD. Orlova AA. Zaytsev VP. Nikitina EV. Peregudov AS. Gurbanov AV. Borisov RS. Khrustalev VN. Maharramov AM. Varlamov AV. Tetrahedron 2009; 65: 3789
- 19 Voskressensky LG. Listratova AV. Borisova TN. Kovaleva SA. Borisov RS. Varlamov AV. Tetrahedron 2008; 64: 10443
- 20 Voskressensky LG. Kulikova LN. Gozun SV. Khrustalev VN. Borisova TN. Listratova AV. Ovcharov MV. Varlamov AV. Tetrahedron Lett. 2011; 52: 4189
- 21 Voskressensky LG. Borisov RS. Kulikova LN. Kleimenov AV. Borisova TN. Varlamov AV. Chem. Heterocycl. Compd. 2012; 48: 680
- 22 Cho H. Iwama Y. Sugimoto K. Kwon E. Tokuyama H. Heterocycles 2009; 78: 1183
- 23 Cho H. Iwama Y. Sugimoto K. Mori S. Tokuyama H. J. Org. Chem. 2010; 75: 627
- 24 Cho H. Tetrahedron 2014; 70: 3527
- 25 Cho H. Iwama Y. Okano K. Tokuyama H. Synlett 2013; 24: 813
- 26 González-Gómez Á. Domónguez G. Pérez-Castells J. Tetrahedron 2009; 65: 3378
- 27 Zhang L. Chang L. Hu H. Wang H. Yao Z.-J. Wang S. Chem. Eur. J. 2014; 20: 2925
- 28 Harthong S. Bach R. Besnard C. Guénée L. Lacour J. Synthesis 2013; 45: 2070
- 29 Mohammadizadeh MR. Alborz M. Tetrahedron Lett. 2014; 55: 6808
- 30 Malkova AV. Polyanskii KB. Soldatenkov AT. Soldatova SA. Merkulova NL. Khrustalev VN. Russ. J. Org. Chem. 2016; 52: 219
- 31 Majumdar KC. Chattopadhyay B. Samanta S. Tetrahedron Lett. 2009; 50: 3178
- 32 Majumdar KC. Samanta S. Chattopadhyay B. Tetrahedron Lett. 2009; 50: 4866
- 33 Majumdar KC. Samanta S. Chattopadhyay B. Nandi RK. Synthesis 2010; 985
- 34 Majumdar KC. Mondal S. Ghosh D. Chattopadhyay B. Synthesis 2010; 1315
- 35 Majumdar KC. Ghosh T. Chakravorty S. Tetrahedron Lett. 2010; 51: 3372
- 36 Lee HS. Kim SH. Kim TH. Kim JN. Tetrahedron Lett. 2008; 49: 1773
- 37 Lee HS. Kim SH. Gowrisankar S. Kim JN. Tetrahedron 2008; 64: 7183
- 38 Sunderhaus JD. Dockendorff C. Martin SF. Tetrahedron 2009; 65: 6454
- 39 Bennasar M.-L. Zulaica E. Solé D. Alonso S. Tetrahedron 2012; 68: 4641
- 40 Sahn JJ. Martin SF. Tetrahedron Lett. 2011; 52: 6855
- 41 Jäger M. Görls H. Günther W. Schubert US. Chem. Eur. J. 2013; 19: 2150
- 42 Peshkov AA. Peshkov VA. Pereshivko OP. Van Hecke K. Kumar R. Van der Eycken EV. J. Org. Chem. 2015; 80: 6598
- 43 Yu RT. Friedman RK. Rovis T. J. Am. Chem. Soc. 2009; 131: 13250
- 44 Koya S. Yamanoi K. Yamasaki R. Azumaya I. Masu H. Saito S. Org. Lett. 2009; 11: 5438
- 45 Kumar P. Zhang K. Louie J. Angew. Chem. Int. Ed. 2012; 51: 8602
- 46 Thakur A. Facer ME. Louie J. Angew. Chem. Int. Ed. 2013; 52: 12161
- 47 Shaw MH. Croft RA. Whittingham WG. Bower JF. J. Am. Chem. Soc. 2015; 137: 8054
- 48 Hassam M. More SV. Li W.-S. Mendeleev Commun. 2014; 24: 159
- 49 Pavlyuk O. Teller H. McMills MC. Tetrahedron Lett. 2009; 50: 2716
- 50 Wood K. Black DSt. C. Namboothiri IN. N. Kumar N. Tetrahedron Lett. 2010; 51: 1606
- 51 Wood K. Black DSt. C. Kumar N. Tetrahedron 2011; 67: 4093
- 52 Majumdar KC. Mondal S. Ghosh D. Synthesis 2010; 1176
- 53 Majumdar KC. Samanta S. Chattopadhyay B. Nandi RK. Synthesis 2010; 863
- 54 Fadeyi OO. Senter TJ. Hahn KN. Lindsley CW. Chem. Eur. J. 2012; 18: 5826
- 55 Senter TJ. Schulte ML. Konkol LC. Wadzinski TE. Lindsley CW. Tetrahedron Lett. 2013; 54: 1645
- 56 Benedetti E. Lomazzi M. Tibiletti F. Goddard J.-P. Fensterbank L. Malacria M. Palmisano G. Penoni A. Synthesis 2012; 44: 3523
- 57 Curto JM. Kozlowski MC. J. Org. Chem. 2014; 79: 5359
- 58 Dhara K. Midya GC. Dash J. J. Org. Chem. 2012; 77: 8071
- 59 Moss TA. Tetrahedron Lett. 2013; 54: 4254
- 60 Jagadeesh Y. Ramakrishna K. Rao BV. Tetrahedron: Asymmetry 2012; 23: 697
- 61 Sletten EM. Bertozzi CR. Org. Lett. 2008; 10: 3097
- 62 Mak XY. Crombie AL. Danheiser RL. J. Org. Chem. 2011; 76: 1852
- 63 Willumstad TP. Haze O. Mak XY. Lam TY. Wang Y.-P. Danheiser RL. J. Org. Chem. 2013; 78: 11450
- 64 Bennasar M.-L. Zulaica E. Solé D. Roca T. García-Díaz D. Alonso S. J. Org. Chem. 2009; 74: 8359
- 65 Bennasar M.-L. Solé D. Zulaica E. Alonso S. Tetrahedron 2013; 69: 2534
- 66 Nilson MG. Funk RL. Org. Lett. 2010; 12: 4912
- 67 Brahma K. Achari B. Chowdhury C. Synthesis 2013; 45: 545
- 68 Putey A. Popowycz F. Do Q.-T. Bernard P. Talapatra SK. Kozielski F. Galmarini CM. Joseph B. J. Med. Chem. 2009; 52: 5916
- 69 Blaszykowski C. Aktoudianakis E. Alberico D. Bressy C. Hulcoop DG. Jafarpour F. Joushaghani A. Laleu B. Lautens M. J. Org. Chem. 2008; 73: 1888
- 70 Donets PA. Van Hecke K. Van Meervelt LE. Van der Eycken V. Org. Lett. 2009; 11: 3618
- 71 Peshkov VA. Van Hove S. Donets PA. Pereshivko OP. Van Hecke K. Van Meervelt L. Van der Eycken EV. Eur. J. Org. Chem. 2011; 1837
- 72 Kumar A. Li Z. Sharma SK. Parmar VS. Van der Eycken EV. Chem. Commun. 2013; 49: 6803
- 73 Modha SG. Vachhani DD. Jacobs J. Van Meervelt L. Van der Eycken EV. Chem. Commun. 2012; 48: 6550
- 74 Alcaide B. Almendros P. Cembellín S. del Campo TM. J. Org. Chem. 2015; 80: 4650
- 75 Tokimizu Y. Oishi S. Fujii N. Ohno H. Org. Lett. 2014; 16: 3138
- 76 Ferrer C. Escribano-Cuesta A. Echavarren AM. Tetrahedron 2009; 65: 9015
- 77 Majumdar KC. Mondal S. Ghosh D. Tetrahedron Lett. 2010; 51: 348
- 78 Zhang J. Wang Y.-Q. Wang X.-W. Li W.-DZ. J. Org. Chem. 2013; 78: 6154
- 79 Diaba F. Martínez-Laporta A. Bonjoch J. J. Org. Chem. 2014; 79: 9365
- 80 Fang X. Liu K. Li C. J. Am. Chem. Soc. 2010; 132: 2274
- 81 Cincinelli R. Dallavalle S. Merlini L. Nannei R. Scaglioni L. Tetrahedron 2009; 65: 3465
- 82 Pandey G. Kant R. Batra S. Tetrahedron Lett. 2015; 56: 930
- 83 Seo S.-Y. Kim G. Tetrahedron Lett. 2015; 56: 3835
- 84 Patir S. Uludag N. Tetrahedron 2009; 65: 115
- 85 Yokosaka T. Nemoto T. Hamada Y. Chem. Commun. 2012; 48: 5431
- 86 Yokosaka T. Kanehira T. Nakayama H. Nemoto T. Hamada Y. Tetrahedron 2014; 70: 2151
- 87 Martin CL. Overman LE. Rohde JM. J. Am. Chem. Soc. 2010; 132: 4894
- 88 Falcon BV. Creighton CJ. Parker MH. Reitz AB. Synth. Commun. 2008; 38: 411
- 89 Ranatunga S. Tang C.-HA. Kang CW. Kriss CL. Kloppenburg BJ. Hu C.-CA. Del Valle JR. J. Med. Chem. 2014; 57: 4289
- 90 Alcaide B. Almendros P. Aragoncillo C. Fernandez I. Gomez-Campillos G. J. Org. Chem. 2014; 79: 7075
- 91 Bariwal JB. Ermolat’ev DS. Glasnov TN. Van Hecke K. Mehta VP. Van Meervelt L. Kappe CO. Van der Eycken EV. Org. Lett. 2010; 12: 2774
- 92 Cheung LL. W. Yudin AK. Org. Lett. 2009; 11: 1281
- 93 Cheung LL. W. Yudin AK. Chem. Eur. J. 2010; 16: 4100
- 94 Smith MR. Blake AJ. Hayes CJ. Stevens MF. G. Moody CJ. J. Org. Chem. 2009; 74: 9372
- 95 Mukhina OA. Kumar NN. B. Cowge TM. Kutateladze AG. J. Org. Chem. 2014; 79: 10956
- 96 Nakamura I. Sato Y. Takeda K. Terada M. Chem. Eur. J. 2014; 20: 10214
- 97 Zhao C. Xie X. Duan S. Li H. Fang R. She X. Angew. Chem. Int. Ed. 2014; 53: 10789
- 98 Feng S. Wang Z. Zhang W. Xie X. She X. Chem. Asian J. 2016; 11: 2167
- 99 Anand A. Singh P. Mehra V. Amandeep, Kumar V. Mahajan MP. Tetrahedron Lett. 2012; 53: 2417
- 100 Granger BA. Jewett IT. Butler JD. Martin SF. Tetrahedron 2014; 70: 4094
- 101 Huo H.-H. Zhang H.-K. Xia X.-E. Huang P.-Q. Org. Lett. 2012; 14: 4834
- 102 Huo H.-H. Xia X.-E. Zhang H.-K. Huang P.-Q. J. Org. Chem. 2013; 78: 455
- 103 Uludag N. Yılmaz R. Asutay O. Colak N. Chem. Heterocycl. Compd. 2016; 52: 196
- 104 Hehir S. O’Donovan L. Carty MP. Aldabbagh F. Tetrahedron 2008; 64: 4196
- 105 Poloukhtine A. Rassadin V. Kuzmin A. Popik VV. J. Org. Chem. 2010; 75: 5953
- 106 Prasad B. Adepu R. Sharma AK. Pal M. Chem. Commun. 2015; 51: 1259
- 107 Yavari I. Seyfi S. Synlett 2012; 23: 1209
- 108 Patil VP. Ghosh A. Sonavane U. Joshi R. Sawant R. Jadhav S. Waghmode SB. Tetrahedron: Asymmetry 2014; 25: 489
- 109 Liu R.-R. Li B.-L. Lu J. Shen C. Gao J.-R. Jia Y.-X. J. Am. Chem. Soc. 2016; 138: 5198
- 110 Xu R. Xia R. Luo M. Xu X. Cheng J. Shao X. Li Z. J. Agric. Food Chem. 2014; 62: 381
- 111 Aron ZD. Ito T. May TL. Overman LE. Wang J. J. Org. Chem. 2013; 78: 9929
- 112 Gong X. Chen M. Yao L. Wu J. Chem. Asian. J. 2016; 11: 1613
- 113 Boeckman RK. Jr. Genung NE. Chen K. Ryder TR. Org. Lett. 2010; 12: 1628
- 114 Daidone G. Sprio V. Plescia S. Marino ML. Bombieri G. Bruno G. J. Chem. Soc., Perkin Trans. 2 1989; 137
- 115 Maggio B. Raffa D. Raimondi MV. Cusimano MG. Plescia F. Cascioferro S. Cancemi G. Lauricella M. Carlisi D. Daidone G. Eur. J. Med. Chem. 2014; 72: 1
- 116 Vicente J. Saura-Llamas I. Garcıa-López J.-A. Bautista D. Organometallics 2010; 29: 4320
- 117 Frutos-Pedreno R. Gonzalez-Herrero P. Vicente J. Organometallics 2012; 31: 3361
- 118 García-Lopez J.-A. Oliva-Madrid M.-J. Saura-Llamas I. Bautista D. Vicente J. Organometallics 2012; 31: 6351
- 119 Oliva-Madrid M.-J. García-Lopez J.-A. Saura-Llamas I. Bautista D. Vicente J. Organometallics 2014; 33: 6420