

RSS-Feed abonnieren
DOI: 10.1055/s-0040-1720032
Synthetic Approaches to Diospongins: A Two Decade Journey
Authors
The author Dr. Krishnaji acknowledges CHRIST (Deemed to be University) for funding though the Major Research Project (MRP # MRPDSC-1723).

Abstract
Tetrahydropyran units having multiple stereogenic centers serve as excellent building blocks for various active pharmaceutical ingredients (APIs). In particular, the presence of the unique molecular architecture of the trisubstituted tetrahydropyran (THP) unit in diospongins enhances their biological activity due to multiple stereogenic centers and has attracted attention from the synthetic community over the last two decades. In this review, we discuss synthetic approaches to chiral and racemic forms of diospongins during the period 2006–2020 in chronological order.
Key words
diospongins - diarylheptanoids - tetrahydropyran - cross-metathesis - hetero-Diels–Alder reaction - asymmetric allylation - enzyme resolution - oxa-Michael addition - Prins cyclizationBiographical Sketch

Dr. Krishnaji Tadiparthi was born in Eluru, Andhra Pradesh, India, and obtained his Ph.D. in 2008 from the Indian Institute of Chemical Technology (IICT), Hyderabad with Dr. Ahmed Kamal. He carried out postdoctoral studies at the Freie University Berlin, Germany (with Prof. Hans-Ulrich Reissig) and Versailles Saint-Quentin-en-Yvelines University, France (with Prof. Gwilherm Evano). He has held positions as Senior Scientist at AstraZeneca, Bangalore, and Senior Manager at Dr. Reddy’s Laboratory, Hyderabad. Currently, he is an Assistant Professor in the department of chemistry, CHRIST (Deemed to be University), Bangalore, India. His research interests are in the areas of organic synthesis, including the development of novel synthetic transformations and total synthesis of biologically active molecules using bio-catalytic principles.

Sourav Chatterjee was born in Burnpur, West Bengal, India and received his B.Sc. (hons) (Chemistry) from Kazi Nazrul University, West Bengal. Currently he is pursuing his M.Sc. (Organic Chemistry) from CHRIST (Deemed to be University), Bangalore and is carrying out research on the development of novel synthetic transformations in the group of Dr. Krishnaji Tadiparthi.
Introduction
Oxygen-containing heterocyclic compounds, especially tetrahydrofurans and tetrahydropyrans, are important scaffolds in many biologically active compounds.[1] [2] [3] [4] Diospongins belong to a family of cyclic 1,7-diarylheptanoids isolated from rhizomes of Dioscorea spongiosa in 2004 by Kadota et al. Diospongins have 2,6-cis- and 2,6-trans-tetrahydro-2H-pyran rings, as a core, constructed by intramolecular cyclization of 5,7-dihydroxy-1,7-diphenyl-2-hepten-1-one, in their biosynthesis. Moreover, these compounds possess a trisubstituted tetrahydropyran unit with different stereochemistries at C-3.
Diospongins A and B exhibit inhibitory activity against bone resorption induced by parathyroid hormone in a bone organ culture, with diospongin B showing more potent anti-osteoporotic activity than diospongin A, presumably due to the different configurations of their tetrahydropyran rings.[5] [6] [7] [8]

In a continuation of our efforts towards the synthesis of various biologically active molecules,[3d] [9] [10] we present the synthetic efforts that have been made toward the synthesis of various diospongins from 2006 to date in the form of a review in chronological order (Figure [1]). Moreover, this review should help the synthetic community to explore further the synthesis of various trisubstituted tetrahydropyran-containing natural products (Figure [2]). Most of the synthetic approaches reported so far have been asymmetric syntheses, but enantioselective approaches involving chiral auxiliaries, chiral pool precursors, or resolution have also been reported. In summary, the key steps involved in the racemic and chiral synthetic approaches, including the total number of steps and overall yields, are collected and compared in Table [1].

Synthesis of Chiral Diospongins
Chandrasekhar’s Approach (2005)
In 2005, Chandrasekhar et al. reported the synthesis of (–)-diospongin A (1) using a Keck asymmetric allylation, a base-catalysed conjugate addition of an α,β-unsaturated ester, and an intramolecular oxy-Michael reaction as the key steps.[11] Initially, the optically pure allyl phenyl carbinol 9, was subjected to one-pot ozonolysis followed by Wittig olefination to provide the α,β-unsaturated ester 10 in 79% yield. Next, the protected syn-1,3-diol derivative 11 was obtained by base-catalysed intramolecular conjugate addition of PhCHO with 10 in the presence of tBuOK in 61% yield. The ester group in 11 was then reduced with LiAlH4 to furnish compound 12 in 77% yield. The (E)-enone 13 was obtained from 12 by one-pot IBX-mediated oxidation followed by Wittig olefination in 77% yield over two steps. Finally, (–)-diospongin A (1) was obtained from 13 by hydrolysis of the benzylidene acetal group and intramolecular oxy-Michael addition in 69% overall yield (Scheme [1]).

Cossy’s Approach (2006)
In 2006, Bressy and Cossy reported the synthesis of (–)-diospongin A (1) using two enantioselective allyl titanations, cross-metathesis, and an intramolecular oxy-Michael reaction as key steps.[12] Initially, optically pure allylic alcohol 9 (ee > 98%) was prepared from benzaldehyde 14 by reaction with allyl titanium complex (R,R)-Ti. The hydroxyl group was protected as its TBS ether 15 in 75% yield. Next, the terminal olefin in 15 was subjected to oxidative cleavage to obtain the corresponding aldehyde, which was then treated in situ with the highly face-selective complex (R,R)-Ti to obtain the 1,3-syn-diol 16 (dr 95:5, 87% yield for two steps). Next, 1,3-diol 16 was subjected to cross-metathesis with phenyl vinyl ketone using Grubbs’ catalyst (G-II) to afford the 1,7-diarylheptenone 17 (75%, E/Z95:5). Finally, (–)-diospongin A (1) was obtained in 60% yield from 17 by intramolecular oxy-Michael addition using TBAF (Scheme [2]).

Jennings’ Approach (2006)
In 2006, Sawant and Jennings demonstrated the synthesis of (–)-diospongins A 1 and B 2 using stereoselective reduction of an oxocarbenium cation as a key step.[13] Initially, alcohol 9 was subjected to esterification to furnish the dienic ester 18 in 68% yield. Next, compound 18, upon ring-closing olefin metathesis using Grubbs’ catalyst (G-II), provided the unsaturated lactone 19 in 90% yield. Subsequently, 19, upon epoxidation in the presence of hydroperoxide, afforded the epoxy lactone 20 stereoselectively in 85% yield. The oxirane was converted into TES ether 22 by regioselective reduction followed by hydroxyl group protection in 89% overall yield. Next, the lactone 22 was reduced to the corresponding lactol using DIBAL-H, followed by acetylation with Ac2O to provide 23. Finally, (–)-diospongin B (2) was obtained by reaction of 23 with BF3·OEt2 to form the corresponding oxocarbenium cation, followed by concomitant removal of the TES group in 81% yield (Scheme [3]).

This approach was also modified to allow the preparation of diospongin A.[14] Lactone 21 was reacted with allyl magnesium bromide to obtain the corresponding lactol, which was treated with TFA in situ to afford the oxocarbenium species followed by reduction with Et3SiH to furnish TES ether 22.[15] Subsequently, compound 22 was transformed into aldehyde 23 by reductive ozonolysis in 95% yield. Next, Grignard addition to the aldehyde provided the secondary alcohol, followed by Dess–Martin periodinane oxidation to give ketone 24 in 91% yield for two steps. Finally, (–)-diospongin A (1) was obtained in 85% yield by deprotection of the TES ether 24 with 5% HCl (Scheme [4]).

Uenishi’s Approach (2007)
In 2011, Uenishi’s group executed the synthesis of (–)-diospongins A (1) and B (2) using CBS reduction, Brown allylation, and Wacker oxidation as key steps.[14] The synthesis started from compound 25, which was subjected to Brown allylation using (+)-Ipc2B-allyl, to furnish 26a in 62% yield along with diastereoisomer 26b in 14% yield. Next, compound 26a was converted into 27 by ozonolysis, followed by Wittig olefination in 80% yield over two steps. Subsequently, the hydroxyl group in 27 was protected as a TBS ether 28 in 86% yield. The reduction of the keto-group in 28 with (R)-CBS (98%, 85% de), followed by deprotection of the silyl ether, provided triol 29a in 94% yield. Next, compound 29a was subjected to cyclization using PdCl2(CH3CN)2, to furnish cis-(E)-tetrahydropyran 30 in 92% yield. Finally, (–)-diospongin B (2) was obtained from 30 by MOM protection, Wacker oxidation, followed by removal of the MOM group with aq. HCl in 91% yield. Similarly, (–)-diospongin A (1) was obtained from 28 by reduction of the keto-group with (S)-CBS, TBS deprotection followed by cyclization and Wacker oxidation in moderate overall yield (Scheme [5]).

Yadav’s Approach (2007)
In 2007, our group reported the synthesis of (–)-diospongin A (1) by using Prins cyclization and enzymatic kinetic resolution as key steps.[16] Initially, the homoallylic alcohol 9 was subjected to Prins cyclization with cinnamaldehyde 31 to form 32 in 78% yield as a single diastereomer. Next, 32 was subjected to enzymatic resolution using porcine pancreatic lipase (PPL) to produce acetate 33 and alcohol 32a. Next, acetate 33 was hydrolysed using K2CO3 to provide alcohol 32b in 92% yield (94% ee). Inversion of the alcohol 32b was achieved under Mitsunobu conditions to lead to 34 in 90% yield. Finally, (–)-diospongin A (1) was obtained by Wacker oxidation followed by hydrolysis of 34 in 90% overall yield (Scheme [6]).

Bates’ Approach (2007)
In 2007, Bates and Song executed the synthesis of (–)-diospongin A (1) using cross-metathesis, and intramolecular Michael addition as key steps.[17] Initially, (S)-phenylbutenol 9 was converted into tert-butyl carbonate 35. Next, compound 35 was converted into epoxy alcohol 37 by iodocyclization, followed by methanolysis using potassium carbonate. The hydroxyl group in 37 was protected as its TBS ether 38 and subsequent ring-opening of the epoxide with vinyl magnesium bromide and CuBr to provide homoallylic alcohol 39. Finally, (–)-diospongin A (1) was obtained by cross-metathesis of 39 with phenyl vinyl ketone in the presence of Grubbs’ catalyst (G-II), followed by one-pot TBS deprotection and cyclization using Amberlyst 15 in 83% yield (Scheme [7]).

Sabitha′s approach (2008)
In 2008, our group demonstrated the synthesis of (–)-diospongins A 1 and B 2 using Keck allylation, stereoselective reduction, Horner–Wadsworth–Emmons olefination, and intramolecular oxy-Michael reaction as key steps.[18] The synthesis commenced with the chiral 1-phenyl but-3-en-1-ol 9, prepared from benzaldehyde by Keck allylation with allyl tributyltin in the presence of (S)-BINOL and Ti(OiPr)4 (73%, 97% ee). Oxidative cleavage of the terminal olefin moiety of 9 produced the corresponding aldehyde, which, without isolation, was treated with ethyl diazoacetate in the presence of a catalytic amount of tin(II) chloride to afford the β-keto ester 40 in 80% yield. The syn-selective reduction of δ-hydroxy-β-keto ester 40 was performed with catecholborane to afford the syn-1,3-diol 41 in 75% yield. Cyclization of ester 41 was accomplished with p-TsOH to afford δ-lactone 42 in 68% yield. Next, reduction of lactone 42 with DIBAL-H followed by Horner–Wittig olefination provided (–)-diospongin A (1) and (–)-diospongin B (2) (4:6 ratio) in 60% yield over two steps (Scheme [8]).

Xian’s approach (2008)
In 2008, Xian et al. demonstrated syntheses of diospongin A and B by using Linchpin coupling, Luchi reduction, and Mitsunobu inversion as key steps.[15] Initially, the Linchpin coupling of TBS dithiane 43 with epoxides (+)-44 and (+)-45 provided the alcohol (–)-46 in 74% yield. Next, compound (+)-46was subjected to dithiane deprotection, Dess–Martin oxidation, and acidic cyclization to furnish the dihydropyranone (+)-47a. Subsequently, compound (+)-47awas subjected to Luche reduction followed by hydrogenation, and benzyl group deprotection to afford the diol (–)-48a, either in one pot or stepwise. Next, the primary hydroxyl group in (–)-48a was oxidized with TEMPO/NaClO2, followed by reaction with PhMgBr to give 49a. Then, selective oxidation of the benzylic hydroxyl of 49a using Dess–Martin periodinane followed by Mitsunobu inversion completed the synthesis of (–)-diospongin A (1) (Scheme [9]).

Synthesis of (–)-diospongin B (2) was initiated by switching the order of epoxide addition with 43 under the same protocol to afford (–)-50 in a similar yield to that of the earlier approach. Next, Luche reduction of (+)-47b provided alcohol (+)-50 in high yield. Hydroxyl group-directed hydrogenation of (+)-50 was performed in the presence of chlorotris(triphenylphosphine)rhodium [(Ph3P)3RhCl] to give (+)-51a in modest yield along with stereoisomer (+)-51b as by-product. Finally, the synthesis of (–)-diospongin B (2) was completed in four steps from (+)-51a (Scheme [10]).

Kumaraswamy’s Approach (2009)
In 2009, Kumaraswamy et al. demonstrated the enantioselective synthesis of (–)-diospongin A (1) and ent-diospongin A (3) from achiral starting materials.[19] The synthesis commenced with the asymmetric hetero-Diels–Alder reaction between Danishefsky’s diene 50 and furfuraldehyde 51 in the presence of (S)-BINOL/Ti(OiPr)4 to furnish dihydropyranone 52 (60%, 99.9% ee). Next, compound 52 was treated with phenylboronic acid in the presence of Rh(cod)2BF4 to obtain the 1,4-addition product 53 in a 95% yield. Reduction of the ketone in 53 was performed with Noyori’s catalyst, (R,R-diamine-Ru catalyst A) to afford alcohol 54 (96%, >99.9% de). The hydroxyl group in 54 was protected as its PMB ether 55 in 98% yield. Next, the furyl group of 55 was subjected to oxidative cleavage followed by esterification and DIBAL-H reduction to provide 56 in 88% yield over three steps. Aldehyde 56 was subjected to Horner–Emmons olefination and subsequent hydrolysis of the intermediate enol ether to provide 57 in 75% yield. Finally, 57 was converted into (–)-diospongin B (2) using DDQ in 92% yield. Furthermore, the C-5 hydroxyl group of (–)-diospongin B (2) was protected as its TBDPS ether to furnish 58. Unexpectedly, deprotection of the TBDPS group of 58 with excess TBAF furnished (–)-diospongin A (1) in 86% yield (Scheme [11]). Similarly, ent-diospongin A (3) was obtained from the hetero-Diels–Alder reaction between Danishefsky’s diene 50 and furfuraldehyde 51 in the presence of (R)-BINOL/Ti(OiPr)4 followed by a similar sequence of reactions (Scheme [12]).


Hashimoto’s Approach (2010)
In 2010, Hashimoto et al. demonstrated the preparation of (–)-diospongin A 1 and B 2 using sequential enantioselective hetero-Diels–Alder and TMSOTf-catalysed Mukaiyama–Michael reactions as a key steps.[20] Initially the HDA reaction between Danishefsky’s diene (53) and benzaldehyde (14) was performed in the presence of [Rh2(S-BPTPI)4] and subsequently the reaction mixture was treated with TMSOTf at –78 °C to obtain the dihydropyranone 56. Next, the reaction mixture was treated with silyl enol ether X to provide 54 (85%, 95% ee). Finally, (–)-diospongin B (2) was obtained by chemo- and stereoselective reduction of 54 with K-Selectride as a single diastereomer in 86% yield. Furthermore, (–)-diospongin A (1) was obtained from (–)-diospongin B (2) in 89% yield by using 30% hydrochloric acid (Scheme [13]).

Meshram’s Approach (2011)
In 2011, Kumar and Meshram executed the synthesis of (–)-diospongin A (1) using the enantioselective Mukaiyama aldol reaction, diastereoselective reduction of δ-hydroxy-β-keto ester, and intramolecular oxa-Michael reaction as key steps.[21] The synthesis started with stereoselective Mukaiyama aldol reaction between Chan’s diene 58 and PhCHO 14 in the presence of Ti(OiPr)4/(S)-BINOL (1:1) to obtain the aldol product 59 (81%, >95% ee). Next, δ-hydroxy-β-keto ester 59 was reduced selectively using Zn(BH4)2 to obtain the syn-1,3-diol 60 (87%, syn/anti 10:1). Subsequently, 1,3-syn-diol 60 was protected as its acetonide followed by DIBAL-H reduction and ortho-iodoxybenzoic acid (IBX) oxidation to provide aldehyde 62 in 79% yield over two steps. Finally, (–)-diospongin A (1) was obtained from aldehyde 62 by Wittig olefination and one-pot deprotection–cyclization in the presence of CSA in good overall yield (Scheme [14]).

Piva’s Approach (2011)
In 2011, Piva’s group demonstrated the synthesis of diospongin A homologues using Prins cyclization, Mitsunobu inversion, cross-metathesis, and Wacker oxidation as key steps.[22] Initially, hepta-1,6-dien-4-ol (64), was subjected to Prins cyclization with benzaldehyde in the presence of trifluoroacetic acid, followed by methanolysis to furnish the syn,syn-diastereomer 65 in high yield. Next, cross-metathesis was performed with 65 and styrene in the presence of Grubbs’ catalyst (G-II) to prepare 66 in 68% yield, and 66 was subjected to Wacker oxidation to produce 67a and 68a (Scheme [15]). Similarly, diospongin A homologues were prepared from 70 by Mitsunobu reaction to prepare 71 in 88% yield. A similar set of reactions was performed on unsaturated tetrahydropyranol 71 with styrene cross-metathesis, followed by Wacker oxidation to provide both homologues 67a and 67b (Scheme [16]).


Reddy’s Approach (2011)
In 2011, Reddy et al. synthesized ent-diospongin A (3) by intramolecular oxa-Michael addition, Mitsunobu reaction, and hydrogenation as key steps.[23] The synthesis commenced with the hydroxy-Weinreb amide 74, which could be prepared from l-malic acid in three steps.[24] Weinreb amide 74 was then treated with phenylacetylene in the presence of n-BuLi to form β-hydroxyalkynone 75. Next, compound 75 was converted into dihydropyranone 76 in 85% yield by treating with AgOTf. Compound 76 was subjected to hydrogenation in the presence of palladium on charcoal to form cis-tetrahydropyranol 77, which was subjected to Mitsunobu esterification followed by TBDPS deprotection to give benzoate 79 in 80% yield. Oxidation of the alcohol group in 79 with Dess–Martin periodinane gave the corresponding aldehyde, followed by Wittig reaction to provide tetrahydropyranol 30b (1:9 E/Z). However, hydrolysis of the benzoate ester also occurred during Wittig reaction under the basic conditions. Finally, ent-diospongin A (3) was obtained by Wacker oxidation of 30b in 52% yield (Scheme [17]).

Taylor’s Approach (2013)
In 2013, Taylor’s group demonstrated the synthesis of (–)-diospongins A (1) and B (2) by using second-generation activation conditions for ether transfer as a key step.[25] Accordingly, 2-bromoethoxymethyl ether 80 was converted into syn-1,3-diol mono ether 81 by activating with NIS/1-phenyl-1H-tetrazole-5-thiol, followed by oxidative cleavage with m-CPBA. Next, conjugate addition of 81 to phenyl ketone afforded the vinylogous ester 82 in 85% yield. Finally, (–)-diospongin A (1) was obtained by reductive deprotection of the 2-bromoethyl ether in 75% yield. For the synthesis of (–)-diospongin B (2), compound 83 was prepared from 81 through alkylation with diazomethylsulfonyltoluene in a 66% yield. Next, the sulfonyl pyran was treated with lithium bis(trimethylsilyl)amide followed by treatment with aluminum chloride, in the presence of TBS-enol ether, and zinc-mediated deprotection to obtain (–)-diospongin B (2; Scheme [18]).

Meruva’s Approach (2014)
In 2014, Meruva et al. accomplished the synthesis of ent-diospongin A (3) and epi-diospongin B (6) with Julia–Kocienski olefination, Weinreb amide formation, and Wacker oxidation as key steps.[26] The synthesis commenced from sulfone 84 with Julia–Kocienski olefination with benzaldehyde 14 to obtain olefin ester 85 as a 92:8 mixture of E/Zdiastereomers. This diastereomeric mixture 85 was subjected to ester hydrolysis, followed by formation of Weinreb amide 87. Next, compound 87 was treated with phenyl Grignard reagent to obtain benzoyl derivative 88 in an 85% yield. Reduction of 88 was performed under Luche conditions to provide alcohol 89, followed by acetonide deprotection to give triol 90, which was treated with FeCl3 to furnish cyclized products 30c (40%) and 30d (26%), which were separated by column chromatography. Finally, ent-diospongin A (3) and epi-diospongin B (4) were obtained from 30c and 30d, respectively, by Wacker oxidation (Scheme [19]).

Fall’s Approach (2015)
The synthesis of (–)-diospogin A (1) and ent-diospongin A (3) was demonstrated by Fall et al. from tri-O-acetyl-d-glucal, by using copper-catalysed Michael addition of phenyllitium.[27] Initially, compound 92 was synthesized from 91 in two steps. Next, 92 was subjected to PDC-mediated oxidation to form α,β-unsaturated ketone 93 followed by copper-catalysed Michael addition of PhLi, to give diastereomeric ketones 94a and 94b. The ketone group of 94a was reduced selectively using l-Selectride to give alcohol 95a followed by subsequent protection to give MOM ether 96 in 90% yield. Next, compound 96 was converted into 101a by protection and deprotection steps. Alcohol 101a was converted into nitrile 102a by tosylation followed by tosylate displacement with sodium cyanide in quantitative yield. Nitrile 102a was reduced with DIBAL-H followed by PhLi addition to furnish alcohol 103a in 67% yield over the two steps. Finally, ent-diospongin A (3) was obtained from alcohol 103a by PDC-mediated oxidation followed by MOM deprotection (Scheme [20]). Similarly, (–)-diospongin A (1) was also prepared by using a similar sequence of reactions from ketone 94b (Scheme [21]).


Hall’s Approach (2015)
In 2015, Hall’s group demonstrated the synthesis of (–)-diospongin B using Suzuki–Miyaura cross-coupling and inverse-electron demand oxa [4+2] cycloaddition as key steps.[28] The synthesis started with the preparation of 107 using inverse-electron demand oxa [4+2] cycloaddition in 77% yield (96% ee). Next, selective epoxidation of 108 was performed in the presence of m-CPBA, to form 109 in a 70% yield (13:1). The epoxide ring was opened with DIBAL-H followed by hydroxyl group protection as its TES ether 111 in 72% yield for two steps. Finally, synthesis of (–)-diospongin B (2) was completed by Mukaiyama-type addition of 112 with 111 followed by TES group removal in 66% yield over two steps (Scheme [22]).

Clarke’s Approach (2016)
In 2016, Clarke et al. demonstrated the synthesis of (–)-diospongin B (2) using the Maitland–Japp reaction as a key step.[29] Accordingly, the synthesis commenced with dihydropyran formation 114 using the Maitland–Japp reaction in the presence of the dimethylacetal of N,N-dimethyl formamide (97%), followed by conjugate addition of Ph2CuLi to give 115 in 91% yield. Next, decarboxylation was performed under microwave conditions to provide the required tetrahydropyran-4-one, which was further reduced with l-Selectride to form THP 30c as the major diastereomer (9:1). Finally, (–)-diospongin B (2) was obtained from 116 by MOM protection and Wacker oxidation followed by MOM deprotection (Scheme [23]).

Prasad’s Approach (2020)
In 2020, Vaithegi and Prasad executed the synthesis of (–)-diospongin A isomers using vinylogous a Mukaiyama aldol reaction and oxa-Michael addition reaction as key steps.[30] The synthetic sequence commenced with the Nagao acetate aldol reaction of benzaldehyde with 117 to give aldol products (118a and 118b). Next the β-alkoxy aldehyde 120 was prepared from the aldol product 118a by hydroxyl protection followed by DIBAL-H reduction. Subsequently, Mukaiyama aldol reaction of the aldehyde 120 with the silyl enol ether of allyl phenyl ketone afforded the mono silyloxy protected diol 121 in 71% yield, as an inseparable mixture of diastereomers (77:23) and the diol 122 in 14% yield (79:21). Reaction of the alcohol 121 with camphor sulfonic acid resulted in smooth deprotection of the TES group and this was followed by oxa-Michael reaction, affording (–)-diospongin A (ent-3) in 18% yield and 5-epi-diospongin A (5-epi-ent- 5) in 66% yield. Similarly, the same products were obtained from 122 in the presence of TFA. Furthermore, Mitsunobu inversion of 5-epi-ent- 5 afforded ent-diospongin A (ent-3) in 67% yield (Scheme [24]).

Synthesis of Racemic Diospongins
Piva’s Approach (2007)
In 2007, the Piva group demonstrated the synthesis of (±)-diospongin A (7) using a Prins cyclization and Mitsunobu reaction as key steps.[31] The key intermediate homoallylic alcohol 125 was prepared from benzaldehyde 14 using two allylation reactions, followed by selective oxidation of the benzylic alcohol 124. Next, the homoallylic alcohol 125 was subjected to Prins reaction with benzaldehyde in the presence of TFA to give the tetrahydropyran, which was subjected to hydrolysis to furnish alcohol 126, an epimer of diospongin A, in 83% yield over the two steps. Next, (±)-diospongin A (7) was obtained from 126 using a Mitsunobu inversion in 71% yield over two steps (Scheme [25]).

Hong’s Approach (2009)
In 2009, Hong’s group explored a tandem cross-metathesis and thermal SN2′ approach to prepare (–)-diospongin A (7) without using protecting groups in the synthetic sequence.[32] Initially allyl Grignard reagent was added to benzaldehyde to give the corresponding homoallylic alcohol 125 in 86% yield. Next, alkene 125 was subjected to oxidative cleavage to give the aldehyde and subsequent treatment with allyltrimethylsilane provided 1,3-diols 127 (syn/anti 5:1) that were separated by column chromatography. The tandem cross-metathesis and SN2′ sequence was performed with 1,3-syn-diol 127 and allylbromide using Grubbs’ catalyst (G-II) to afford the 4-hydroxy-2,6-cis-tetrahydropyran 128 (5:1 dr, 83%). Finally, (±)-diospongin A (7) was obtained by a cross-metathesis reaction of 128 with styrene followed by carbonyl group introduction by a Wacker reaction (Scheme [26]).

More’s Approach (2010)
In 2010, More demonstrated the synthesis of (±)-diospongin A (7) using hetero-Diels–Alder reaction and an anchimeric assistance-controlled C-glycosylation as key steps.[33] Accordingly, the [4+2] cycloaddition of diene 129 and benzaldehyde 14 was performed in the presence of achiral chromium catalyst D to provide 130 in 79% yield. Next, 130 was subjected to Luche reduction to furnish alcohol 131 as a single isomer. The C-5 hydroxyl group was protected as its TBS ether 132, followed by acetoxylation gave 133 as a mixture of anomers (13:1). The TBS group in 133 was removed, followed by esterification under Mitsunobu conditions to give the epimeric benzoate 135. Finally, (±)-diospongin A (7) was obtained from 135 by treating with enol silane C and BF3·OEt2 followed by deacylation in 47% yield over two steps (Scheme [27]).

Gracza’s Approach (2011)
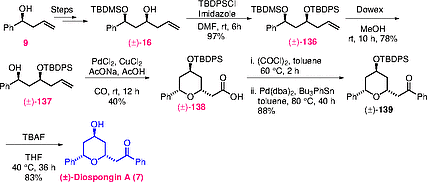
In 2011, Gracza and co-workers explored an approach using palladium (II)-catalysed intramolecular hydroxycarbonylation and Stille coupling as key steps.[34] The key syn-diol 136 was prepared from the homoallylic alcohol 16 by a series of simple protection and deprotection steps. Next, the allylic alcohol was protected as its TBDPS ether, followed by selective cleavage of the TBDMS group with acidic Dowex to afford 137 in 76% yield over two steps. Compound 137 was subjected to hydroxycarbonylation using carbon monoxide in acetic acid, providing exclusively the 2,6-cis-diastereomer 138 in 40% yield. Subsequently, carboxylic acid 138 was converted into (±)-diospongin A (7) in a three-step sequence (Scheme [28]).
Ho’s approach (2012)
In 2012, the group of Ho synthesized (±)-diospongin A (7) by desymmetric cyclization and reduction of a meso-1,7-diarylheptanoid as key steps.[35] Initially, ditosylate 141 was prepared from 140 in 36% yield. Next, the two hydroxyl groups were protected as the benzaldehyde dimethyl acetal to give 142 in a 77% yield. After this, compound 142 was treated with 2-lithio-2-phenyl-1,3-dithiane to give 143 in 78% yield. Finally, (±)-diospongin A (7) was obtained from 143 by debenzylidenation, desulfurization, and cyclization (Scheme [29]).

Tong’s Approach (2013)
In 2013, Tong’s group demonstrated the synthesis of (±)-diospongin A (7) using an efficient 4-step [3+2+1] strategy.[36] Initially allylic alcohol 146 was converted into α,β-unsaturated isoxazoline 147 through a three-step sequence of Dess–Martin oxidation, oxime formation, and [3+2]-cycloaddition with styrene. Next, isoxazoline 147 was subjected to chemoselective ring-opening with SmI2, followed by intramolecular 6-endo-trig-oxa-Michael cyclization to obtain tetrahydropyran-4-one 148. Finally, (±)-diospongin A (7) was obtained from 148 by desilylation, Dess–Martin oxidation and chemoselective reduction with K-selectride in 20% overall yield (Scheme [30]).

Gharpure’s Approach (2015)
In 2015, the Gharpure group executed the synthesis of (±)-diospongin B using a stereoselective intramolecular cyclopropanation of a vinylogous carbonate with carbene using a copper catalyst as the key step.[37] The approach commenced with selective protection of the primary alcohol of butane-1,3-diol (149), as its TBS ether, followed by addition of the secondary alcohol to ethyl propiolate using N-methyl morpholine (NMM), to provide the vinylogous carbonate 150. Next 150 was converted into acid 151 using Jones’ reagent by simultaneous protodesilylation, followed by oxidation, in 68% overall yield. Subsequently, acid 151 was converted into diazo ketone 152 by reaction with oxalyl chloride, followed by reaction with diazomethane, in 41% yield over two steps. Compound 152 was then converted into donor-acceptor cyclopropane 153, the regioselective ring-opening reaction of which was performed in the presence of n-Bu3SnH and AIBN to give pyranone 154. The keto group in 154 was selectively reduced with l-Selectride to obtain the alcohol 155 (d.r. 19:1), followed by TBS protection to afford ester 156. Finally, (±)-diospongin B (8) was obtained by converting the ester into the Weinreb amide, phenyl Grignard addition, and deprotection of TBS (Scheme [31]).

Conclusions and Outlook
In conclusion, we have compiled and presented all the synthetic approaches to optically active and racemic diospongins from 2006 to date. All the 26 synthetic approaches have been described in chronological order. Having compared the synthetic routes, we conclude that the routes of Hashimoto (2010)[20] and Jennings (2006)[13] are most efficient in terms of yields and number of steps to furnish chiral diospongin derivatives. However, Piva’s approach (2007)[31] seems to be best as a racemic approach.
Conflict of Interest
The author declares no conflict of interest.
-
References
- 1a Perron F, Albizati KM. Chem. Rev. 1989; 89: 1617
- 1b Shimizu YM. Chem. Rev. 1993; 93: 1685
- 1c Yasumoto T, Murata M. Chem. Rev. 1993; 93: 1897
- 1d lvarez AA. E, Candenas ML, Perez R, Ravelo JL, Martın JD. Chem. Rev. 1995; 95: 1953
- 2a Elliott MC. Contemp. Org. Synth. 1994; 1: 457
- 2b Burns CJ, Middleton DS. Contemp. Org. Synth. 1996; 3: 229
- 2c Elliott MC. Contemp. Org. Synth. 1997; 4: 238
- 2d Elliott MC. J. Chem. Soc., Perkin Trans. 1 1998; 4175
- 2e Elliott MC. J. Chem. Soc., Perkin Trans. 1 2000; 1291
- 2f Elliott MC, Williams E. J. Chem. Soc., Perkin Trans. 1 2001; 2303
- 2g Elliott MC. J. Chem. Soc., Perkin Trans. 1 2002; 2301
- 3a Clarke PA, Santos S. Eur. J. Org. Chem. 2006; 2045
- 3b Nasir NMd, Ermanis K, Clark PA. Org. Biomol. Chem. 2014; 12: 3323
- 3c Fuwa H. Mar. Drugs 2016; 14: 65
- 3d Tadiparthi K. ChemistrySelect 2021; 45: 12702
- 4 For a review on tetrahydrofuran-containing natural products, see: Fernandes RA, Pathare RS, Gorve DA. Chem. Asian J. 2020; 15: 2815
- 5a Zhu J, Islas-Gonzalez G, Bois-Choussy M. Org. Prep. Proced. Int. 2000; 32: 505
- 5b Claeson P, Claeson UP, Tuchinda P, Rentrakul V. Studies in Natural Product Chemistry, Vol. 26. Atta-ur-Rahman, Elsevier; Amsterdam: 2002: 881
- 5c Kadota S, Tezuka Y, Prasaim JK, Ali MS, Banskota AH. Curr. Top. Med. Chem. 2003; 3: 203
- 6a Scapagnini G, Colombrita C, Amadio M, D’Agata V, Arcelli E, Sapienza M, Quattrone A, Calabrese V. Antioxid. Redox Signaling 2006; 8: 395
- 6b Wang Z, Zhang Y, Banerjee S, Li Y, Sarkar FH. Cancer 2006; 106: 2503
- 7a Mohamad H, Lajis NH, Abas F, Ali AM, Sukari MA, Kibazuki H, Nakatani N. J. Nat. Prod. 2005; 68: 285
- 7b Kim J.-H, Lee KW, Lee M.-W, Lee HJ, Kim S.-H, Surh Y.-J. FEBS Lett. 2006; 580: 385
- 8 Yin J, Kouda K, Tezuka Y, Le Tran Q, Miyahara T, Chen Y, Kadota S. Planta Med. 2004; 70: 54
- 9a Tadiparthi K, Anand P, Sakirolla R, Gupta TP, Jadhav KA, Das SK, Yadav JS. Chem. Rec. 2022; 22: 60
- 9b Tadiparthi K, Roy A, Sakirolla R, Reddy BV. S, Yadav JS. Chem. Rec. 2022; e202200044
- 9c Tadiparthi K, Venkatesh S. J. Heterocycl. Chem. 2022; doi.org/10.1002/jhet.4480
- 9d Tadiparthi K, Anand P. Org. Process Res. Dev. 2021; 25: 2008
- 9e Tadiparthi K, Devaroy MJ. L, Panigrahi A, Sakirolla R. Asian J. Org. Chem. 2021; 8: 2084
- 9f Tadiparthi K, Anand P. Synthesis 2021; 53: 3469
- 10a Raghavendra S, Tadiparthi K, Yadav JS. Carbohydr. Res. 2017; 442: 17
- 10b Kamal A, Khanna GB. R, Krishnaji T. Helv. Chim. Acta 2007; 90: 1723
- 10c Tadiparthi K, Kamal A, Raghavendra S. Asian J. Chem. 2017; 29: 2321
- 10d Kamal A, Krishnaji T, Reddy PV. Tetrahedron: Asymmetry 2007; 18: 1775
- 10e Kamal A, Krishnaji T, Reddy PV. Tetrahedron Lett. 2007; 48: 7232
- 10f Kamal A, Krishnaji T, Khanna GB. R. Tetrahedron Lett. 2006; 47: 8657
- 11 Chandrasekhar S, Shyamsunder T, Prakash SJ, Prabhakar A, Jagadeesh B. Tetrahedron Lett. 2006; 47: 47 ; In this paper the product was named ‘(–)-diospongin B’; however, this isomer was subsequently referred to as ‘(–)-diospongin A’ in subsequent publications (compound 1 in this review)
- 12 Bressy C, Allais F, Cossy J. Synlett 2006; 3455
- 13 Sawant KB, Jennings M. J. Org. Chem. 2006; 71: 7911
- 14 Kawai N, Hande SM, Uenishi J. Tetrahedron 2007; 63: 9049
- 15 Wang H, Shuhler BT, Xian M. Synlett 2008; 2651
- 16 Yadav J, Padmavani B, Reddy BV. S, Venugopal Ch, Rao AB. Synlett 2007; 2045
- 17 Bates R, Song P. Tetrahedron Lett. 2007; 63: 4497
- 18 Sabitha G, Padmaja P, Yadav JS. Helv. Chim. Acta 2008; 91: 2235
- 19 Kumaraswamy G, Ramakrishna G, Naresh P, Jagadeesh B, Sridhar B. J. Org. Chem. 2009; 74: 8468
- 20 Anada M, Washio T, Watanabe Y, Takeda K, Hashimoto S. Eur. J. Org. Chem. 2010; 6850
- 21 Kumar RN, Meshram HM. Tetrahedron Lett. 2011; 52: 1003
- 22 Raffier L, Izquierdo F, Piva O. Synthesis 2011; 4037
- 23 Reddy ChR, Reddy GB, Srikanth B. Tetrahedron: Asymmetry 2011; 22: 1725
- 24 Shioiri T, McFarlane N, Hamada Y. Heterocycles 1998; 47: 73
- 25 Stefan E, Nalin AP, Taylor RE. Tetrahedron 2013; 69: 7706
- 26 Meruva SB, Mekala R, Raghunadh A, Rao KR, Dahanukar VH, Pratap TV, Kumar UK. S, Dubey PK. Tetrahedron Lett. 2014; 55: 4739
- 27 Zúñiga A, Pérez M, Gándara Z, Fall A, Gómez G, Fall Y. ARKIVOC 2015; (vii): 195
- 28 Rybak T, Hall DG. Org. Lett. 2015; 17: 4165
- 29 Clarke PA, Nasir NM, Sellars PB, Peter AM, Lawson CA, Burroughs JL. Org. Biomol. Chem. 2016; 14: 6840
- 30 Vaithegi K, Prasad KR. Tetrahedron 2020; 76: 131625
- 31 Hiebel M.-A, Pelotier B, Piva O. Tetrahedron 2007; 63: 7874
- 32 Lee K, Kim H, Hong J. Org. Lett. 2009; 11: 5202
- 33 More JD. Synthesis 2010; 2419
- 34 Karlubíkov O, Babjak M, Gracza T. Tetrahedron 2011; 67: 4980
- 35 Ho T.-L, Tang B, Ma G, Xu P. J. Chin. Chem. Soc. 2012; 59: 455
- 36 Yao H, Ren J, Tong R. Chem. Commun. 2013; 49: 193
- 37 Gharpure SJ, Mane SP, Nanda LN, Shukla MK. Isr. J. Chem. 2016; 56: 553
For reviews on oxygen-containing natural products, see:
For a series of review articles on the synthesis of saturated oxygen heterocycles, see:
For reviews on synthesis of tetrahydropyran derivatives, see:
Isolation and biological activity of diospongin:
Corresponding Author
Publikationsverlauf
Eingereicht: 09. Juni 2022
Angenommen nach Revision: 20. Juni 2022
Artikel online veröffentlicht:
14. Juli 2022
© 2022. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1a Perron F, Albizati KM. Chem. Rev. 1989; 89: 1617
- 1b Shimizu YM. Chem. Rev. 1993; 93: 1685
- 1c Yasumoto T, Murata M. Chem. Rev. 1993; 93: 1897
- 1d lvarez AA. E, Candenas ML, Perez R, Ravelo JL, Martın JD. Chem. Rev. 1995; 95: 1953
- 2a Elliott MC. Contemp. Org. Synth. 1994; 1: 457
- 2b Burns CJ, Middleton DS. Contemp. Org. Synth. 1996; 3: 229
- 2c Elliott MC. Contemp. Org. Synth. 1997; 4: 238
- 2d Elliott MC. J. Chem. Soc., Perkin Trans. 1 1998; 4175
- 2e Elliott MC. J. Chem. Soc., Perkin Trans. 1 2000; 1291
- 2f Elliott MC, Williams E. J. Chem. Soc., Perkin Trans. 1 2001; 2303
- 2g Elliott MC. J. Chem. Soc., Perkin Trans. 1 2002; 2301
- 3a Clarke PA, Santos S. Eur. J. Org. Chem. 2006; 2045
- 3b Nasir NMd, Ermanis K, Clark PA. Org. Biomol. Chem. 2014; 12: 3323
- 3c Fuwa H. Mar. Drugs 2016; 14: 65
- 3d Tadiparthi K. ChemistrySelect 2021; 45: 12702
- 4 For a review on tetrahydrofuran-containing natural products, see: Fernandes RA, Pathare RS, Gorve DA. Chem. Asian J. 2020; 15: 2815
- 5a Zhu J, Islas-Gonzalez G, Bois-Choussy M. Org. Prep. Proced. Int. 2000; 32: 505
- 5b Claeson P, Claeson UP, Tuchinda P, Rentrakul V. Studies in Natural Product Chemistry, Vol. 26. Atta-ur-Rahman, Elsevier; Amsterdam: 2002: 881
- 5c Kadota S, Tezuka Y, Prasaim JK, Ali MS, Banskota AH. Curr. Top. Med. Chem. 2003; 3: 203
- 6a Scapagnini G, Colombrita C, Amadio M, D’Agata V, Arcelli E, Sapienza M, Quattrone A, Calabrese V. Antioxid. Redox Signaling 2006; 8: 395
- 6b Wang Z, Zhang Y, Banerjee S, Li Y, Sarkar FH. Cancer 2006; 106: 2503
- 7a Mohamad H, Lajis NH, Abas F, Ali AM, Sukari MA, Kibazuki H, Nakatani N. J. Nat. Prod. 2005; 68: 285
- 7b Kim J.-H, Lee KW, Lee M.-W, Lee HJ, Kim S.-H, Surh Y.-J. FEBS Lett. 2006; 580: 385
- 8 Yin J, Kouda K, Tezuka Y, Le Tran Q, Miyahara T, Chen Y, Kadota S. Planta Med. 2004; 70: 54
- 9a Tadiparthi K, Anand P, Sakirolla R, Gupta TP, Jadhav KA, Das SK, Yadav JS. Chem. Rec. 2022; 22: 60
- 9b Tadiparthi K, Roy A, Sakirolla R, Reddy BV. S, Yadav JS. Chem. Rec. 2022; e202200044
- 9c Tadiparthi K, Venkatesh S. J. Heterocycl. Chem. 2022; doi.org/10.1002/jhet.4480
- 9d Tadiparthi K, Anand P. Org. Process Res. Dev. 2021; 25: 2008
- 9e Tadiparthi K, Devaroy MJ. L, Panigrahi A, Sakirolla R. Asian J. Org. Chem. 2021; 8: 2084
- 9f Tadiparthi K, Anand P. Synthesis 2021; 53: 3469
- 10a Raghavendra S, Tadiparthi K, Yadav JS. Carbohydr. Res. 2017; 442: 17
- 10b Kamal A, Khanna GB. R, Krishnaji T. Helv. Chim. Acta 2007; 90: 1723
- 10c Tadiparthi K, Kamal A, Raghavendra S. Asian J. Chem. 2017; 29: 2321
- 10d Kamal A, Krishnaji T, Reddy PV. Tetrahedron: Asymmetry 2007; 18: 1775
- 10e Kamal A, Krishnaji T, Reddy PV. Tetrahedron Lett. 2007; 48: 7232
- 10f Kamal A, Krishnaji T, Khanna GB. R. Tetrahedron Lett. 2006; 47: 8657
- 11 Chandrasekhar S, Shyamsunder T, Prakash SJ, Prabhakar A, Jagadeesh B. Tetrahedron Lett. 2006; 47: 47 ; In this paper the product was named ‘(–)-diospongin B’; however, this isomer was subsequently referred to as ‘(–)-diospongin A’ in subsequent publications (compound 1 in this review)
- 12 Bressy C, Allais F, Cossy J. Synlett 2006; 3455
- 13 Sawant KB, Jennings M. J. Org. Chem. 2006; 71: 7911
- 14 Kawai N, Hande SM, Uenishi J. Tetrahedron 2007; 63: 9049
- 15 Wang H, Shuhler BT, Xian M. Synlett 2008; 2651
- 16 Yadav J, Padmavani B, Reddy BV. S, Venugopal Ch, Rao AB. Synlett 2007; 2045
- 17 Bates R, Song P. Tetrahedron Lett. 2007; 63: 4497
- 18 Sabitha G, Padmaja P, Yadav JS. Helv. Chim. Acta 2008; 91: 2235
- 19 Kumaraswamy G, Ramakrishna G, Naresh P, Jagadeesh B, Sridhar B. J. Org. Chem. 2009; 74: 8468
- 20 Anada M, Washio T, Watanabe Y, Takeda K, Hashimoto S. Eur. J. Org. Chem. 2010; 6850
- 21 Kumar RN, Meshram HM. Tetrahedron Lett. 2011; 52: 1003
- 22 Raffier L, Izquierdo F, Piva O. Synthesis 2011; 4037
- 23 Reddy ChR, Reddy GB, Srikanth B. Tetrahedron: Asymmetry 2011; 22: 1725
- 24 Shioiri T, McFarlane N, Hamada Y. Heterocycles 1998; 47: 73
- 25 Stefan E, Nalin AP, Taylor RE. Tetrahedron 2013; 69: 7706
- 26 Meruva SB, Mekala R, Raghunadh A, Rao KR, Dahanukar VH, Pratap TV, Kumar UK. S, Dubey PK. Tetrahedron Lett. 2014; 55: 4739
- 27 Zúñiga A, Pérez M, Gándara Z, Fall A, Gómez G, Fall Y. ARKIVOC 2015; (vii): 195
- 28 Rybak T, Hall DG. Org. Lett. 2015; 17: 4165
- 29 Clarke PA, Nasir NM, Sellars PB, Peter AM, Lawson CA, Burroughs JL. Org. Biomol. Chem. 2016; 14: 6840
- 30 Vaithegi K, Prasad KR. Tetrahedron 2020; 76: 131625
- 31 Hiebel M.-A, Pelotier B, Piva O. Tetrahedron 2007; 63: 7874
- 32 Lee K, Kim H, Hong J. Org. Lett. 2009; 11: 5202
- 33 More JD. Synthesis 2010; 2419
- 34 Karlubíkov O, Babjak M, Gracza T. Tetrahedron 2011; 67: 4980
- 35 Ho T.-L, Tang B, Ma G, Xu P. J. Chin. Chem. Soc. 2012; 59: 455
- 36 Yao H, Ren J, Tong R. Chem. Commun. 2013; 49: 193
- 37 Gharpure SJ, Mane SP, Nanda LN, Shukla MK. Isr. J. Chem. 2016; 56: 553
For reviews on oxygen-containing natural products, see:
For a series of review articles on the synthesis of saturated oxygen heterocycles, see:
For reviews on synthesis of tetrahydropyran derivatives, see:
Isolation and biological activity of diospongin: