

Subscribe to RSS
DOI: 10.1055/s-0040-1720125
A Review of Recent Progress on the Anticancer Activity of Heterocyclic Compounds
Authors
B.N. is thankful to Basic Scientific Research, University Grants Commission (BSR UGC) for start up grant No. F.30-358/2017(BSR).

Abstract
Cancer is one of the most daunting illnesses in the world as compared to many other human diseases. This review article aims to summarize the literature that is already published based on heterocyclic anticancer compounds. Under this broad topic we try to shed a light on anticancer potentiality of oxygen-, sulfur-, and nitrogen-containing heterocyclic compounds, such as quinolines, pyrroles, pyrimidines, pyridines, indoles, also sulfonamides linked heterocycles, benzimidazoles and oxadiazoles.
1 Introduction
1.1 Drugs in Use for Cancer Treatment
1.2 Recently Discovered Anticancer Drugs
2 Various Classes of Compounds as Anticancer Agents
2.1 Quinoline Derivatives as Anticancer Agents
2.2 Benzimidazoles as Anticancer Agents
2.3 Indole: A Privileged Scaffold for the Design of Anticancer Agents
2.4 Pyrimidine Derivatives as Anticancer Agents
2.5 Pyridine Derivatives as Anticancer Agents
2.6 Pyrrole Derivatives as Anticancer Agents
2.7 Sulfonamides linked with heterocycles as Anticancer Agents
2.8 Oxadiazole and Its Derivatives as Anticancer Compounds
2.9 Benzothiazole-Triazole Hybrids as Anticancer Compounds
3 Conclusion
Biographical Sketches

Dr Beena Negi is an alumna of Gargi college and presently is a lecturer at Gargi college, University of Delhi (UoD). She is a trained synthetic organic chemist. She has published numerous research papers in international journals. She has authored a book on the chemistry of heterocyclic compounds. Her academic interests are green chemistry and synthetic medicinal chemistry. She was the deputy coordinator for Open Book Examinations, UoD. She was a member of the editorial team of the annual magazine of UoD Highlights 2021, 2022, and 2023. She had been a member of committee of courses for both UG and PG at the Department of Chemistry, University of Delhi. Her lectures on green chemistry are available on her you tube channel https://www.youtube.com/watch?v=6sGDo6UW89w&t=18s.

Aarshiya Kwatra has completed her Bachelor’s degree from Miranda House, University of Delhi followed by a Master’s degree from M.D.U., Rohtak. She did her B.Ed. from M.D.U. Rohtak and has mentored many children for IIT-JEE and NEET. Her academic interests are Green Chemistry and Medicinal Chemistry.
Introduction
Cancer is a disorder caused by the uncontrolled proliferation of cells which affects the surrounding tissues.[1] Many anticancer medications have been developed and are available in the market but the need to produce anticancer drugs persists because the current drugs are poisonous, inefficient, less soluble, and less selective.[2] The World Health Organisation (WHO) has developed a worldwide action plan for 2013–2030 to prevent and control noncommunicable diseases with the goal of reducing premature death from cancer,[3] cardiovascular disease, diabetes, and chronic respiratory diseases by 25%.[4] This review article compiles all available literature on the cytotoxic action of various chemicals on cancer cells. Scientists, researchers, scholars, and manufacturers have worked extensively on therapeutic targets and development tactics.
1.1Drugs in Use for Cancer Treatment
Anticancer agents are difficult to classify; previously they were grouped into six categories: (a) alkylating agents, (b) antimetabolites, (c) natural products, (d) hormones and antagonists, (e) miscellaneous. However, several of the most important agents have recently joined the miscellaneous group.
Alkylating chemicals and related compounds prevent DNA replication by forming covalent bonds with the SDNA. Antimetabolites are compounds that hinder or interfere with one or more of the metabolic processes responsible for DNA production. Cytotoxic antibiotics or those derived from microbes that inhibit mammalian cell division. Plant compounds such as taxanes, campothecins and vinca alkaloids; the bulk of them have a specific effect on microtubule function, which influences mitotic spindle formation.
The most important hormones include steroids (glucocorticoids, estrogens, and androgens) as well as drugs that limit hormone release or interfere with their effects.[5] Table [1] displays some anticancer agents with different mechanisms of action.
1.2
Recently Discovered Anticancer Drugs
Anticancer medications have many limitations including resistance by chemicals in cancer cells, numerous adverse effects, and the fact that they affect both cancer and healthy cells. The study to produce good anticancer treatments that can overcome the challenges mentioned above is ongoing and a few anticancer drugs are being licensed by the Food and Drug Administration in 2023 (Table [2]).
Heterocyclic compounds exhibit diverse pharmacological activities.[8] Hormones, vitamins, antibiotics, and other biological compounds found in living organisms are composed of heterocyclic molecules. While heterocyclic compounds with a sulfur atom account for the bulk of FDA-approved drugs, heterocyclic compounds with a nitrogen atom are regarded to be the most common type of chemical material utilized in medicinal chemistry.[9] Many physiologically active natural products used in conventional treatment or approved prescription medications contain nitrogen-containing heterocyclic compounds. Many of their synthetic equivalents are available in the therapeutic market to treat a range of ailments. This article discusses the anticancer activity of numerous heterocyclic compounds and derivatives as well as their structures in vitro activity and IC50 values.
|
Entry |
Drug name |
Type of cancer treated |
|
1 |
retifanlimab-dlwr[6] |
metastatic or recurrent locally advanced Merkel cell carcinoma |
|
2 |
enfortumab[7] |
locally advanced or metastatic urothelial carcinoma |
|
3 |
epcoritamab-bysp[6] |
relapsed or refractory diffuse large β-cell lymphoma |
|
4 |
glofitamab-gxbm[6] |
relapsed or refractory diffuse large β-cell lymphoma |
|
5 |
talquetamab-tgv[6] |
relapsed or refractory multiple myeloma |
This review focuses on the anticancer activity of heterocyclic-based compounds.
2
Various Classes of Compounds as Anticancer Agents
2.1Quinoline Derivatives as Anticancer Agents
The bioactive heterocyclic compounds quinoline and quinolone derivatives are important classes in the pharmaceutical field because of their diverse pharmacological properties which include antibacterial, anti-inflammatory, anticancer, anti-HCV, antitubercular, antimalarial, anti-HIV, and anti-Alzheimer activities.[10] Dimers typically exhibited some unusual features when compared to the comparable monomeric compounds which is why they have generated a lot of interest recently. Derivatives of quinoline/quinolone possess good anticancer activity. The following are drugs undergoing clinical trials or are used to treat cancer: quinoline derivative dactolisib (a quinoline dimer); topotecan; vosaroxin (or voreloxin, a quinolone derivative), the first anticancer drug that was approved by the US FDA in 2009 as an orphan drug for treating acute myeloid leukemia; and quarfloxacin (Figure [1]). In addition, results of phase I clinical trial for dactolisib, which is a PI3K inhibitor, displayed its good efficiency in treating a number of solid tumors. So, quinoline/quinolone compounds have the potential to be cancer-fighting substances.
Various review articles and books have made a comprehensive review of current developments of quinoline-based anticancer agents.[16]

Li et al. investigated the in vitro antitumor effects of a series of bisquinoline derivatives connected by the 4-oxy-3-fluoroaniline moiety on a panel of cancer cell lines (H460, HT-29, MKN-45, U87MG, and SMMC-7721) (see Figure [2]).[11] Preliminary data indicate that all compounds tested had moderate to good cytotoxic effects (IC50: 0.011–3.56 μM) on cancer cells with potencies in the single-digit μM range and high selectivity for the H460 and MKN-45 cell lines. Compound 2 was more powerful than its analogue counterpart 1, implying that using fluoro in the R2 position could boost activity. Compound 1b with an IC50 of 0.011–0.15 μM was shown to be 2.9–10.9 times more efficient than foretinib for all selected cancer cell lines highlighting the need for additional study. Its capacity to inhibit c-Met kinase was found to be comparable to foretinib. Further research found that the 2-quinolone can substitute for the quinoline motif. Compared to foretinib, the 2-quinolone-quinoline derivative 3 was shown to be 6.1 times more active against H460, 2.4 times more active against HT-29, and 2.1 times more active against U87MG cell lines with an IC50 value of 0.031–0.52 μM. Compound 3 had significant potency against c-Met (IC50 = 1.21 nM), moderate inhibitory impact against F1t-3 (IC50 = 2.15 nM), and low potency against c-Kit, VEGFR-2, PDGFR-b, and EGFR (IC50 = 362. 8 to>100,000 nM). Based on this research, it can be stated that compound 3 largely affects c-Met and Flt-3 and it may be considered for optimization. [1,2,4]Triazolo[3,4-b][1,3,4]thiadiazole-tethered fluoroquinolone-fluoroquinolone compounds 4 and 5 were tested on L1210, CHO, and HL60 cell lines. All compounds displayed cytotoxicity with IC50 values ranging from 0.12–26.2 μM. Compound 4 with an IC50 value of 0.54 μM and compound 5 with and IC50 value of 0.12 μM had the strongest effects on HL60 cells, proving that the cyclopropyl group of at N1 of fluoroquinolone is crucial for its high cytotoxicity.


Secosteroid-Quinoline Hybrids
13,17-Secoestra-1,3,5(10)-trien-17-oic acid N′-(quinolylmethylene)hydrazides 6 and 7 were shown to have a high selectivity index (Figure [3]).[12] In luciferase reporter assays, compounds 6 and 7 displayed excellent antiestrogenic potential as compared to tamoxifen. Compound 6 and 7 displayed good activity against MCF-7 cells with an IC50 value of 1.7 μM for compound 6 and 1.5 μM for compound 7. NCI/ADR-RES cells are multidrug resistant cells and compounds 6 and 7 were persistent towards these cells with IC50 value 1.5 μM for 6 and 3.5 μM for 7. These were the defining characteristics of these successful drugs.
2.1.2
8-Hydroxyquinoline-Chalcone Hybrids with Dual Tubulin/EGFR Kinase Inhibition

Compounds 8 and 9 were tested for cell cycle, wnt1/β-catenin gene suppression, and apoptosis (Figure [4]).[13] Western blot determined the apoptotic markers Bax, Bcl2, Casp3, Casp9, PARP1, and β-actin. The compounds 8 and 9 are promising antiproliferative candidates that inhibit EGFR kinase and polymerize tubulin. Compounds 8 and 9 exhibited good activity with an IC50 value of 0.097 ± 0.006 μM and 0.334 ± 0.002 μM, respectively against EGFR kinase inhibition and 5.3 ± 0.33 μM and 10.84 ± 0.67 μM, respectively, against tubulin polymerization inhibition
2.1.3
Quinoline Derivatives as Small Molecule Mutant EGFR Inhibitors Targeting Resistance in NSCLC
The selected cell lines were HCC827, H1975 (L858R/T790 M), A549 (WT EGFR), A-549, and BEAS-2B.[14] Many quinoline compounds displayed great cytotoxicity. Among all compounds, 10 (Figure [5]) was found to possess high activity with IC50 values of 0.010 μM, 0.21 μM, 0.99 μM, and 2.99 μM as compared to osimertinib with IC50 values of 0.0042 μM, 0.04 μM, 0.92 μM, and 2.67 μM.

2.1.4
Tetra-, Penta-, and Hexacyclic Phenothiazines Modified with a Quinoline Moiety
Compound 11, a 3-(dimethylamino)propyl-substituted diquinothiazine, showed high activity with an IC50 value of 0.3 μM against A549 cell line (see Figure [6]).[15] Compound 12, a 2-(diethylamino)ethyl-substituted diquinothiazine, displayed good cytotoxic activity against the SNB-19 cell line. Compound 13, possessing a 2-[(2-chloroethyl)ureido]ethyl substituent, has excellent activity with an IC50 value of 87 nM against the SK-MEL-5 melanoma cell line. Leukemia (CCRF-CEM and MOLT4), colon (HCT-116), CNS (SNB-75 and SF-295), prostate (PC-3), non-small cell lung (NCI-H460 and HOP-92), ovary (IGROV-1, OVCAR-4, and OVCAR-5), and breast (MDA-MB-460) were the selected cancer cell lines. Less actions (IC50 = 0.25–1.0 μM) were seen for these cancer lines. 2-(Diethylamino)ethyl-substituted 14 (IC50 = 0.19 μM) and 3-(dimethylamino)propyl-substituted 15 (IC50 = 0.29 μM) displayed good activity against the ovarian cancer cell line IGROV-1.

2.1.5
3,5-Disubstituted Quinolines
3,5-Disubstituted quinolines with substitution at the 3,5-positions are powerful inhibitors of c-Jun N-terminal kinases (JNKs) that produce anticancer action. [16] JNK1, JNK2, and JNK3 are three different genes that produce JNKs. The prospective therapeutic target for neurodegenerative illnesses, such as Parkinson’s disease, Alzheimer’s disease, and other CNS disorders, is JNK3 inhibition because it has been demonstrated to trigger neuronal death. A growing number of medicines for anticancer activities are now focusing on JNK3.[16] Jiang et al. (2007) reported synthesis of a novel family of powerful c-Jun N-terminal kinase (JNK) inhibitors with p38 selectivity.[17] 3,5-Disubstituted quinolines were produced from 4-(2,7-phenanthrolin-9-yl)phenol. The inverse sulfonamide tert-butyl analogue 16 (Figure [7]) in the X-ray crystal structure of JNK3 indicated an unanticipated binding mode for this novel scaffold with the protein and had a strong inhibitory effect against JNK3 (0.15 μM IC50) but not against p38.

2.1.6
4,7-Disubstituted Quinolines
7-Chloro-4-quinolinylhydrazone derivatives 17 (Figure [7]) as anticancer medications was reported by Bispo et al.[18] The synthesized compounds were tested against cancer cell lines using MTT assay. The anticancer activity results demonstrated that compounds had good cytotoxic activity against cancer cell lines, i.e., SF-295, colon; HTC-8 colon, and HL-60 leukemia, with IC50 values in range from 0.314 to 4.65 μg/mL.
Ferrer et al. in 2009 reported many novel [(7-chloroquinolin-4-yl)amino]-substituted chalcone derivatives that were tested for in vitro antiproliferative efficacy against human prostate LNCaP tumor cells (Figure [7]).[19] Compounds 4-chloro-4′-[(7-chloroquinolin-4-yl)amino]chalcone (18) with IC50 value 7.93 ± 2.05 μg/mL, 4′-[(7-chloroquinolin-4-yl)amino]-3-fluorochalcone (19) with IC50 values of 7.11 ± 2.06 μg/mL, and 4′-[(7-chloroquinolin-4-yl)amino]chalcone (20) with IC50 value of 6.95 ± 1.62 μg/mL were good inhibitors of human prostate cell. Xiang et al. reported that 5-methoxy-2-(4-methyl-1,4-diazepan-1-yl)-N-(1-methylpiperidin-4-yl)quinolin-4-amine (21) inhibited EZH2 with an IC50 value of 1.2 μM.[20] Compound 21 reduced H3K27me3 levels across cells and shown effective inhibitory properties for the tumor cell lines HCT15 and MD-MBA-231.
2.1.7
2-Substituted 4-Amino-6-haloquinolines

Jiang et al. designed, synthesized and evaluated series of 2-substituted 4-amino-6-haloquinolines and the cancer cell lines selected were H-460, HT-29, HepG2, and SCG-7901.[21] (E)-N 1-[6-Chloro-2-(4-methoxystyryl)quinoline-4-yl]-N 2,N 2-dimethylethane-1,2-diamine (22) (Figure [8]) has a 4-methoxystyryl group at C2 and a (dimethylamino)alkylamino substituent at C4 has been considered strongly for further structural modifications. This compound possesses good anticancer properties with IC50 values of 0.03 μM against H-460 cancer cell line, 0.55 μM against HepG2 cancer line, and 1.24 μM against SGC-7901 cancer cell line; these values showed that it is 2.5 to 186 more active than gefitinib.
2.1.8
4-Anilino-8-hydroxy-2-phenylquinolines
Chen et al. in 2006 synthesized many 4-anilino-8-methoxy-2-phenylquinoline and 4-anilino-8-hydroxy-2-phenylquinoline derivatives and tested their antiproliferative ability.[22] Compound 23 (Figure [8]) inhibited the development of cancer cells, such as HCT116 (colon cancer) with GI50 = 0.07 μM, MCF7 with GI50 = ˂0.01 μM, and MDA-MB-435 (breast cancer) with GI50 = 0.01 μM.
2.1.9
Indolo, Pyrrolo, Benzofuro, and 6-Anilinoindolo Quinolines
Chen et al. in 2002 prepared indolo-, pyrrolo-, and benzofuro-quinolin-2(1H)-ones and 6-anilinoindoloquinoline derivatives.[23] These were tested in vitro using three cell lines: MCF7 (breast), NCI-H460 (lung), and SF-268 (CNS). 1-[3-(11H-Indolo[3,2-c]quinolin-6-ylamino)phenyl]ethenone oxime hydrochloride (24) with GI50 = 1.70 μM and its 2-chloro derivative 25 with GI50 = 1.35 μM were found to be very potent (Figure [8]). Both these compounds 24 and 25 with a GI50 value of less than 0.01 μM inhibited the growth of SNB-75, which is a CNS cancer cell.
2.2
Benzimidazoles as Anticancer Agents
Benzimidazoles are heterocyclic compounds of great importance. They possess antitumor, antiproliferative, anticancer, antimicrobial, and antioxidant activity.[24] Benzimidazole is structural analogue of purines which are part of DNA nucleotides.[25] Thiabendazole (or thiabenzazole), telmisartan, pimobendan, pantoprazole, omeprazole, etonitazene, carbendazim, benomyl, and albendazole are benzimidazole compounds which possess great pharmacological value. Veliparib, selumetinib, pracinostat, nocodazol, liarozol, glasdegib, galeterona, dovitinib, crenolanib, bendamustina, and abemaciclib are some organic derivatives of benzimidazole and these compounds are in clinical trials for determining their anticancer properties.
De, Banerjee, and co-workers reviewed benzimidazoles in 2023 with the aim to develop a comprehensive SAR (structure–activity relationship).[26] While Ebenezer and co-workers reviewed benzimidazoles in drug discovery between 2020–2022.[27]
Garuti et al. synthesized BZ-4,7-dione compounds (Figure [9]). These compounds were tested against molt-3 cells. These cells were derived from human lymphoblastic leukemia.[28] Compounds 26 (IC50 = 1.32 μM) and 27 (IC50 = 2.63 μM) showed good antiproliferative activity. Compound 28, IC50 = 0.98 μM against SW620 cells, was active compared to doxorubicin (IC50 = 0.72 μM. Chojnacki et al. determined antiproliferative and protein-kinase inhibitory effects in anticancer treatment.[29] Compound 29 with Ki = 1.96 μM and compound 30 with Ki = 0.91 μM displayed good protein kinase 2 (CK2) inhibitory activity. Due to this these compounds can be considered for further research.

Abd El-Meguid et al.[30] reported that compounds 31 (IC50 = 0.25 ± 0.003 μM) and 32 (IC50 = 0.19 ± 0.004 μM) showed good activity as compared to erlotinib (IC50 = 1.23 ± 0.005 μM) against HER2 cell (Figure [10]). Also compound 31 with an IC50 value of 0.157 ± 0.007 μM and compound 32 (IC50 = 0.109 ± 0.014 μM) displayed excellent activity against EGFR when compared to erlotinib (IC50 = 0.079 ± 0.001 μM).

Compound 33 (Figure [10]) possess an IC50 of 4.30 nM against PARP1 and 1.58 nM against PARP2 enzymes substantial in vitro antitumor activity.[31]
Compounds 34 and 35 (Figure [10]) were more active as compared to standard compound staurosporine.[32] Compound 34 possess an IC50 of 2.02 ± 0.13 μM against the MCF-7 cell line and compound 35 displayed an IC50 value of 3.30 ± 0.21 μM against the MDA-MB-231 cell line.
Compound 36 with an IC50 value of 0.33 μM and compound 37 with an IC50 value of 0.38 μM against EGFR kinase where as erlotinib caused inhibition with an IC50 value of 0.39 μM (Figure [11]).[33]


MBIC, dovitinib, selimetinib, and abemaciclib (Figure [12]) are some benzimidazole based anticancer drugs (Figure [12]). Benzimidazole derivatives are good microtubule inhibitor.[34] Methyl 2-(5-fluoro-2-hydroxyphenyl)-1H-benzimidazole-5-carboxylate (MBIC) with a methyl ester is substituted with a hydroxyl group at C-2 whereas a fluoro group at C5 position in the aryl ring inhibits microtubule and are very active against breast cancer cells.[35] MBIC has the following values against: breast cancer cells IC50 0.73–20.4 μmol/L, cervical cancer cells IC50 = 0.21 μmol/L, hepatocellular carcinoma cells IC50 = 2.92 μmol/L, lung cancer cells IC50 = 1.29 μmol/L, and colorectal cancer cells IC50 = 2.72 μmol/L.
Compound 38 is an imidazole[1,5-a]pyridine-benzimidazole hybrid (see Figure [13]) which possess good cytotoxic activity against 60 human cancer cell lines.[36] The GI50 values of compound 38 against cell free assay, leukemia cells, breast cancer cells, ovarian cancer cells, melanoma cells, lung cancer cells, colorectal cancer cells, CNS cancer cells, renal cancer cells, and prostate cancer cells are 1.71 μmol/L, 1.47–2.92 μmol/L, 2.17–3.09 μmol/L, 2.05–6.95 μmol/L, 1.39–4.61 μmol/L, 0.42–3.85 μmol/L, 3.63–7.19 μmol/L, 2.97–5.49 μmol/L, 1.67–7.73 μmol/L, and 1.88–2.85 μmol/L, respectively.
Dovitinib, ((3E)-4-amino-5-fluoro-3-[5-(4-methylpiperazine-1-yl)-1,3-dihydrobenzimidazol-2-ylidene]quinoline-2-one also known as CHIR-258 or TKI258) is the lactate salt of a benzimidazole–quinoline compound that function as a multitargeted growth factor receptor kinase inhibitor.[37] The IC50 values against FLT-3, FGFR1, VEGFR3/FLT4, c-Kit, and FGFR3 are 0.001 μmol/L, 0.008 μmol/L, 0.008 μmol/L, and 0.002 μmol/L, 0.009 μmol/L, respectively.
Selumetinib [6-[4-bromo-2-chlorophenyl)amino]-7-fluoro-3-methyl-3H-benzimidazole-5-carboxylic acid (2-hydroxyethoxy)amide] is a MEK inhibitor; it targets MEK1 and MEK2. It binds at MEK1/2 and disrupts the interaction of both ATP and substrate and the assessment of the ERK activation loop.[38] It has an IC50 for MEK1 of 0.014 μmol/L and MEK 2 of 0.53 μmol/L.
Abemaciclib is a CDK inhibitor. It has been approved by FDA to be used as adjuvant with endocrine therapy to treat breast cancers.[39] Abemaciclib has IC50 = 0.002 μmol/L against CDK4 and 0.01 μmol/L against CDK6. Compounds 39 and 40 are RAF kinase inhibitors (Figure [13]). Compound 39 has IC50 = 0.002 mmol/L and 40 has IC50 = 0.014 mmol/L against the B-RAFV600E oncogenic protein;[40] against melanoma cells, compound 39 has 4.6 μmol/L and 40 has IC50 = 2.3 μmol/L.

(2-Chloro-N-(2-p-tolyl-1H-benzimidazole-5-yl)acetamide (41; Figure [13]), a 2-arylbenzimidazole, is an RTK inhibitor for EGFR, VEGFR-2, and PDGFR.[41] The IC50 value of 41 against liver cancer cells is 2 μmol/L.
Compound 42, with a carbamoyl hydrazone substituent, synthesized by Galal et al. is a CHK2 inhibitor (Figure [13]).[42] It inhibits cell cycle progression in ER+ MCF7 breast, HeLa cervical, and HepG2 hepatocellular cancer cell lines. The IC50 values of 42 for CHK2, cervical cells, and breast cancer cells are 0.041 μmol/L, 11.7 μmol/L, and 13.8 μmol/L, respectively.
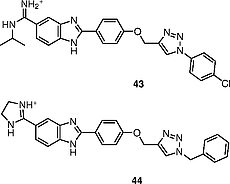
Compounds 43 and 44 (Figure [14]) are 1,2,3-triazolyl-linked 2-arylbenzimidazole derivatives. Compound 43 is a 4-chlorophenyl-substituted 1,2,3-triazolyl N-isopropylamidine whereas compound 44 is a benzyl-substituted 1,2,3-triazolyl imidazoline; both possess excellent activity.[43] Compounds 43 and 44 have IC50 = 0.05 μmol/L and 0.07 μmol/L, respectively, against liver cancer cells.
Rucaparib is a tricyclic benzimidazole carboxamide derivative (Figure [15]). It targets PARP-1, -2, and -3 with IC50 = 0.8 against PARP-1, IC50 = 0.5 against PARP-2, and IC50 = 28 nmol/L against PARP-3.[44] [45] It has been approved by the FDA and the European Medicine Agency for treating ovarian, fallopian tube, and peritoneal cancers as well as advanced solid tumors with evidence of germline or somatic BRCA mutation.

2.3
Indole: A Privileged Scaffold for the Design of Anticancer Agents
Indole and its derivatives have a biologically significant role.[46] It binds to a variety of receptors and enzymes with high affinity because of its distinct structural motif. It is also utilized as anticonvulsant, anti-inflammatory, antimicrobial, antituberculosis, anticancer, anti-HIV, antiviral, and antioxidant. Several synthetic medications that contain indole include sumatriptan, indolemycin, indomethacin, pindolol, and reserpine. Several pharmacologically significant compounds that include indole are: tryptophan, serotonin, indole-3-acetic acid, melatonin, and reserpine.
Wan, Tang, and co-workers have reviewed indole as a privileged scaffold for the design of anticancer agents.[47]
Compound 45 is a 1H-pyrazolo[3,4-b]pyridine derivative (Figure [16]).[48] It possesses good activity for pim kinases inhibition with an IC50 value of 9 nM for pim-1, 39 nM for pim-2, and 12 nM for pim-3. Additionally, compound 45 demonstrated outstanding suppression of additional kinases, including Flt-3, c-Kit, PDGFR, and Kdr. Replacing the 1H-pyrazolo[3,4-b]pyridine core by an indole gives compound 46 (Figure [16]). With an IC50 value of 4 nM for pim-1, 42 nM for pim-2, and 22 nM for pim-3, this substitution preserved the inhibitory activity against pim kinases but lost the ability to inhibit other kinases.

Compound 47 (Figure [17]) displayed good inhibitory activity against pim kinases with an IC50 < 2 nM for pim-1, IC50 = 10 nM for pim-2, and IC50 < 2 nM for pim-3.[48]

An indole alkaloid meridian C (48) was obtained from a marine source (Figure [18]). Its IC50 value of 1.04 μM indicates that it exhibits good action against Pim-1. Lee and co-workers synthesized compound 49 with an IC50 value in the nanomolar range with IC50 values for the kinases Pim-1, Pim-2, and Pim-3 of 3 nM, 110 nM, and 7 nM, respectively.[49] They explored the SAR of 3,5-disubstituted indoles, replacing the 2-aminopyrimidine ring of 49 for a phenyl ring in order to increase its activity. By changing the substituents and locations on the phenyl ring, this was able to maintain the physicochemical properties while observing an increase in hydrophobic contacts with pim-1. As pim inhibitors, these attempts produced a number of unique indole compounds. Compound 50 displayed good activity for pim-1 and pim-3 and its IC50 value was found to be in nanomolar range with single digit value. With the exception of casein kinase 2 (CK2), the most active compound 50 had comparable outstanding inhibitory effects against pim-1 and pim-3 with single digit nanomolar IC50 values and remarkable selectivity over 14 protein kinases. It was significantly more effective than compound 51 in its excellent antiproliferative action against the MV4-11 cell line (IC50 = 0.8 μM). Compound 48 was adapted to increase its inhibitory action against pim-2. Compound 50 displayed three pim isoforms with single digit nanomolar IC50 values (Pim-1: IC50 = 5 nM; Pim-2: IC50 = 259 nM; Pim-3: IC50 = 10 nM).

The FDA or China Food and Drug Administration has approved five HDAC inhibitors: vorinostat, belinostat, romidepsin, panbinostat, and chidamide. Dai and co-workers synthesized and evaluated a series of hydroxamic acid-based HDAC inhibitors with an indole amide residue at the terminus.[50] Compound 52 (Figure [19]) proved to be the most efficacious chemical with a nanomolar IC50 value of 3.1 nM against a mixture of HDACs (mostly HDAC1/2, K562 cell extracts) about 40 times more active than SAHA which had an IC50 value of 143 nM. It showed sub micromolar IC50 values against the human HT1080 cell line (fibrosarcoma; IC50 = 0.12 μM) and the human MDA435 cell line (breast cancer; IC50 = 0.13 μM) which are more than ten times more potent than SAHA.

Giannini et al.[51] designed and synthesized a series of SAHA derivatives bearing the bis(indolyl)methane (BIM) moiety and against the HCT116 and H460 tumor cell lines, compound 53 (Figure [19]) exhibited remarkable antiproliferative properties (IC50 = 0.41 and 0.55 μM, respectively). SAR demonstrated that linker length affects HDAC inhibition with pentamethylene linkers being the most effective. The monoindolyl methane derivatives were not as potent as the target compounds containing the BIM moiety.
2.3.1Novel Indole-Pyrazole Hybrids as Potential Tubulin-Targeting Agents

Compound 54 (Figure [19]) demonstrated a moderate level of inhibition against tubulin polymerization (IC50 = 19 μM) and excellent efficacy against hepatocellular carcinoma (HCC) cell lines with IC50 values in the range of 0.6–2.9 μM.[52]
(E)-3-(2-{[(N-Pentylcarbamimidoyl)hydrazono]methyl}-1H-indol-5-yl)-4-methoxybenzoate (55) possesses and excellent IC50 value of 0.042 μM against SMMC-7721 cells (Figure [20]).[53] However, compound 55 exhibited no cytotoxicity when tested against normal cells, such as HEK293 and HEK293T cells.
2-Phenyl-4,5,6,7-tetrahydro-1H-indole derivative 56 (Figure [20]),[54] with a 4-methoxyphenylhydrazone moiety, is a tubulin polymerization inhibitor and it was tested for its antiproliferative activity in vitro against human breast cancer cell line (MCF-7) and human lung adenocarcinoma cell line (A549). With an IC50 value of 1.77 ± 0.37 μM against MCF-7 and 3.75 ± 0.11 μM against A549, it demonstrated remarkable anticancer activity and had a significant inhibitory effect on tubulin polymerization akin to that of colchicine. Compound 56 exhibited a similar inhibitory effect on tubulin polymerization as colchicine and molecular docking analysis demonstrated that compound 56 possesses a high binding affinity for the tubulin binding pocket of colchicine.[55]
The inhibitory efficacy of compound 57, a sulfonamide derivative of pyridyl-indole-based chalcone (Figure [20]), is an efficient carbonic anhydrase inhibitor; it was tested against the human carbonic anhydrase IX isoform.[56] Studies on fluorescence binding and molecular docking were employed. Utilizing MCF-7 and HepG-2 cell lines, its anticancer efficacy was investigated. Compound 57 with an IC50 value of 0.13 μM, demonstrates remarkable anticancer efficacy.
Li, Li, and co-workers, in 2019, examined indole-imidazole hybrids as powerful tubulin inhibitors and found that 2-(4-methyl-1H-indol-3-yl)imidazole 58 and 2-(1H-indol-4-yl)imidazole 59 (Figure [20]) were very powerful with IC50 = 1.6 to 3.7 nmol/L, which is two to three times greater than the reference drug ABI-231.[57]

Compound 60 is a thiosemicarbazone derivative containing an indole group (Figure [21]).[58] Compound 60 exhibits remarkable anticancer properties as evidenced by its IC50 values of 0.14 ± 0.02 μM for PC3 and 9.85 ± 0.26 μM for WPMY-1. Compound 60 effectively causes apoptosis and inhibits PC3 cell colonization and proliferation. In PC3 cells, compound 60 demonstrated increased selectivity towards two normal WPMY-1 and GES-1.
Ammar, Belal, and co-workers synthesized a novel series of indole derivatives and evaluated their anticancer activity using HepG-2, HCT-116, and MCF-7 cell lines;[59] compound 61 (IC50 <10 μM) demonstrated broad spectrum anticancer action on all three tested cell lines (Figure [21]). Compound 61 exhibits superior antitumor activity compared to lapatinib. Molecular docking was utilized to investigate the binding mechanism, amino acid interactions, and free binding energy of chemical 61.
Compound 62, an indole and pyranoindole derivative possesses good antitumor activity (Figure [21]).[60] Compound 62 demonstrated its inhibitory action on the target protein tubulin and exhibited strong anticancer activity against the HeLa cell line (IC50 = 3.6 ± 0.5). No cytotoxicity was observed by 62 towards normal cells. Data confirmation was achieved using silico assay.
Indole retinoid compound 63 (Figure [21]) exhibits antiproliferative properties in cell lines of liver, breast, and colon cancer.[61] Compound 63 prevents the proliferation of all breast cancer cell lines with extremely low IC50 values. In Huh7 cell line, compound 63 has the highest anticancer activity (IC50 <0.01 μM). Docking and molecular dynamics experiments on RXRα and RXRγ were conducted with compound 63.
The antiproliferative activity of compounds 64 and 65 (Figure [22]), which are 3-thiocyanato-1H-indole derivatives, was assessed against four human cancer cell lines: HL60, HEP-2, NCI-H292, and MCF-7.[62] The reference drug utilized was doxorubicin. Compound 64 antiproliferative activity was measured with IC50 values of 1.43 μM, 2.79 μM, 2.07 μM, and 3.95 μM against HL60, HEP-2, NCI-H292, and MCF-7, respectively. With an IC50 value of 1.10 μM against HL60, 2.80 μM against HEP-2, 2.07 μM against NCI-H292 and 4.05 μM against MCF-7, compound 65 also showed strong antiproliferative activity.

2.4
Pyrimidine Derivatives as Anticancer Agents
It is commonly recognized that pyrimidine derivatives have pharmacological properties. Numerous medications containing pyrimidine group, such as thioguanine, tegafur, and 5-fluorouracil (5-FU) (Figure [23]) were synthesized and employed as anticancer treatments. Pyrimidines and their derivatives have a wide range of biological properties including antibacterial, analgesic, antiviral, and anticancer properties. As a result of the assessment of pyrimidines as a novel class of highly effective anticancer agents, hundreds of compounds, such as 2-cyanopyrimidines, hydrazino pyrimidine-5-carbonitriles, 1,3-dialkylated pyrimidine-2,4-diones, and 4-anilino-2-(2-pyridyl)pyrimidines, have been prepared and evaluated for their anticancer activity.[63] It is acknowledged as the most significant substance in the treatment of cancer because of its structural similarity to the nucleotide base pair found in both DNA and RNA.[64]

Sharma and co-workers reviewed the anticancer activity of pyrimidine with key emphasis on SAR in 2021.[64] Recent advances on pyrimidine derivatives as anticancer agents was reviewed in 2023 by Mahdy, Elnagar, and Sakr.[65]

Pyrimidine-Containing Compounds as Adenosine Receptor Antagonists
In 2022, Li et al. synthesized a number of dual A2A/A2B adenosine receptor (AR) antagonists based on the triazole-pyrimidine-methylbenzonitrile core.[66] Compound 66 (Figure [24]) had remarkable inhibitory effect on A2B AR with an IC50 = 14.12 nM.
2.4.2
Pyrimidine-Containing Compounds as COX-2 Selective Inhibitors
Alam and co-workers synthesized fluorophenyl-substituted cyanopyrimidines in 2020 and assessed them as COX-2 selective inhibitor anticancer medicines.[67] Compound 67 (Figure [24]) demonstrated remarkable anticancer efficacy against ovarian cancer (GI50 = 0.33 μM and selectivity index of 4.84), in contrast to 5-fluorouracil (5-FU) (GI50 = 4.43 μM). Compound 67 was more specific for COX-2 than COX-1 and exhibited broad antitumor action.
2.4.3
Cyanopyrimidines
In 2020, Alam and co-workers synthesized many analogues of benzimidazole-pendant cyanopyrimidine derivatives which were assessed for their in vitro anticancer properties against NCI-60 cancer cell lines at the National Cancer Institute (NCI) in the United States.[68] Of all the compounds produced, compound 68 (Figure [24]) exhibited the highest level of activity.

2.4.4
Disubstituted Pyrimidines
Chen and co-workers, in 2020, synthesized 2,4-disubstituted pyrimidines (Figure [25])[69] and they were tested for antiproliferative activity using MTT assay with VX-680 as positive control against the cell lines A549 (IC50 = 12.05 ± 0.45 μM), HTC-116 (IC50 = 1.31 ± 0.41 μM), and MCF-7 (IC50 = 20.53 ± 6.13 μM); compound 69 exhibited moderate to high levels of activity. Compounds 70 (IC50 = 2.14 to 5.52 μM) and 71 (IC50 = 1.98 to 4.27 μM) differed in their substitution on the aromatic ring and terminal aniline on the pyrimidine moiety and demonstrated excellent activity against the PC-3, A549, MCF-7, and HCT-16 cancer cell lines.[70]
2.4.5
Trisubstituted Pyrimidines
2.4.5.12,4,5-Trisubstituted Pyrimidines
2,4-Diaminopyrimidines are highly effective and specific inhibitors of Aurora A kinase. Using the MTT assay, the compound outstanding activity was evaluated against HeLa, A-549, HCT-8, and Hep-G2 cells and contrasted with VX-680, a positive control. Compound 72 (Figure [26]) demonstrated a cytotoxicity IC50 = 0.5–4.0 μM indicating good activity.[71] HeLa cells in the G2/M phase experience cell cycle arrest due to compound 72.

Cytostatic activities of alkyl 5-alkylpyrimidines, alkyl N-(methoxymethyl)pyrimidines, and 5,6-dihydrofuro[2,3-d]pyrimidines were described (Figure [26]) using 5-FU as the positive control.[72] With an IC50 of 0.8 ± 0.2 mM, 5-(2-chloroethyl)-2,6-dichloropyrimidine (73) had a cytostatic impact on the HCT-116 cancer cell line, resulting in cell cycle arrest at the G2/M phase due to DNA damage. In cellular and enzymatic experiments, 2-(arylamino)pyrimidines with a 2-amino-N-methylbenzamide at C4 and chlorine at C5 shown to be good c-Met inhibitors.[73] Excellent c-Met inhibition was demonstrated by pyrimidines bearing a C2 benzazepinone; the best analogue was compound 74, which had an IC50 value of 10 nM; C3 of the aminobenzamide moiety contains fluorine, which gives c-Met kinase selectivity.
2.4.5.2
2,4,6-Trisubstituted Pyrimidines
2,4,6-Trisubstituted pyrimidine 75 (Figure [27]) demonstrated outstanding efficacy in inhibiting the U937 cell line, with an IC50 value of 12.2 nM. It inhibited the growth of cancer cells by causing polyploidy (4N, 8N, and 16N) by disruption of both chromosomal and spindle formation.[74]

2.4.6
2,4,5,6-Tetrasubstituted Pyrimidines
The MCF-7 cancer cell lines were examined with 2,4-disubstituted pyrimidine derivatives which are ERα and VEGFR-2 ligands. Compound 76 (Figure [27]) demonstrated inhibitory efficacy against VEGFR-2 with an IC50 value of 0.085 μM and an ERα binding affinity of 1.64 μM.[75]
2.4.7
Pyrazolo[1,5-a]pyrimidines and Pyrazolo[4,3-d]pyrimidines
The anticancer activity of the pyrazolo[1,5-a]pyrimidine derivatives with a nitrogen mustard moiety was evaluated using the MTT assay against the cell lines A549, SH-SY5Y, HepG2, MCF-7, and DU145. Compound 77 (Figure [27]) brought the cell cycle to a stop at the G1 phase with an IC50 = 0.2–8.3 μM causing apoptosis in each of the five cancer cell lines.[76]
Pyrazolo[4,3-d]pyrimidines were examined for their anticancer action both in vivo and in vitro and they demonstrated CDK inhibition when the appropriate substitutions were made at C3, C5, and C7.[77] Compound 78 (Figure [28]) demonstrated remarkable inhibition of CDK2, CDK5, and CDK9 with IC50 = 0.002 μM.
Pyrazolo[4,3-d]pyrimidine, a bioisostere of roscovitine, was evaluated for both CDK inhibition and antiproliferative activity. 5-Substituted 3-isopropyl-7-[(4-(2-pyridyl)benzyl)amino]-1(2)H-pyrazolo[4,3-d]pyrimidine 79 (Figure [28]) demonstrated excellent suppression of CDK2 (IC50 = 21 nM) and CDK5 (IC50 = 35 nM).[78]
2.4.8
Pyrrolo[2,3-d]pyrimidines
Derivatives of pyrrolo[2,3-d]pyrimidines exhibit possible inhibitory effects on FAK. Compound 80 (Figure [28]) demonstrated the suppression of MDA-MB231 and A549 cancer cell lines with the IC50 range of 3.20 ± 0.41 to 17.41 ± 1.3 μM, respectively and lowered the FAK enzymatic action at IC50 = 5.4 nM.[79]

When tested against the human cancer cell lines MCF-7 (breast), HCT116 (colorectal), and HepG2 (liver), compound 81 (Figure [28]) demonstrated cytotoxic action.[80] When tested for its ability to inhibit EGFR-TK (epidermal growth factor receptor tyrosine kinase), it demonstrated outstanding inhibitory effects with IC50 values as low as 0.107 μM.
2.5
Pyridine Derivatives as Anticancer Agents
Pyridine is a well-known primary heterocyclic organic compound.[81] Pyridine derivatives have a broad range of biological and medicinal applications, including antiviral,[82] [83] antibacterial,[83]antifungal,[83] anti-inflammatory,[83] anticancer,[83] antineuropathic, antihypertensive,[81] antidiabetic,[83] antitubercular,[83] antiparasitic,[81] antihistaminic,[81] and several other activities. Pyridine-related drugs have high therapeutic properties which persuade medicinal chemists to produce many newly chemotherapeutic agents. Pyridines constitute the class of nitrogenous heterocycles that undergo different chemical synthesis routes for generation of novel compounds possessing anticancer/antitumor properties.[84] Pyridine derivatives are important class of anticancer drug as they possess tendency to inhibit kinases, androgen receptors, tubulin polymerization, topoisomerase, enzyme, human carbonic anhydrase.[85] Sorafenib, regorafenib, vismodegib are pyridine based small molecules that have been approved as anticancer drugs (Figure [29]).
Three relevant reviews appeared in 2022. Alrooqi et al. reviewed pyridine-based heterocyclic compounds as potent anticancer agents from 2017 to 2021.[81] Marinescu and Popa reviewed pyridine compounds with antimicrobial and antiviral activities.[82] Finally De, Kumar S K, and co-workers examined the clinical diversity of the pyridine scaffold.[83]

Pyridine-Ureas as Potential Anticancer Agents

1-[4-Chloro-3-(trifluoromethyl)phenyl]-3-[6-(4-methoxyphenyl)-2-methylpyridin-3-yl]urea (82) (Figure [30]) and 1-[3-(trifluoromethyl)phenyl]-1-(6-(4-methoxyphenyl)-2-methylpyridin-3-yl)urea (83) are two new pyridine-ureas.[86] 3-[3-(Trifluoromethyl)phenyl]urea 83 was tested for its ability to suppress the proliferation of the MCF-7 breast cancer cell line in vitro. Compounds 82 and 83 were assessed for their in vitro anticancer activity utilizing the US-NCI procedure. Comparing compound 82 to the reference medication doxorubicin (IC50 = 1.93 μM), compound 82 is a more potent and essential congener against MCF7 cells (IC50 = 0.22 μM).
2.5.2
Pyridine Derivatives Containing Triazole and Benzimidazole Moieties
A series of N-{4-[2-(1-ammoniaylidyne-4-(3,4,5-trimethoxyphenyl)-1H-furan-2-yl)-3H-imidazo[4,5-b]pyridin-6-yl]phenyl}-2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamide derivatives 84–93 were designed and synthesized (Figure [30]).[87] These compounds anticancer properties were tested against A549 Colon-20, A2780, and MCF-7 and their results were contrasted with those of the reference medication, etoposide. Compounds 85–89 and 93 exhibited remarkable anticancer properties.
2.5.3
Pyridine Derivatives Containing a Pyrazole Moiety
The anticancer activities of pyridine derivatives (Z)-3-amino-7-(4-methoxybenzylidene)-1,7-dihydro-4H-pyrazolo[4,3-c]pyridine-4,6(5H)-dione (94) and (Z)-3-amino-7-(4-chlorobenzylidene)-1,7-dihydro-4H-pyrazolo[4,3-c]pyridine-4,6(5H)-dione (95) were studied (Figure [30]).[88] Comparing compound 94 to the reference medication doxorubicin (IC50 = 4.749 and 2.527 μg/mL, respectively) showed that compound 94 had excellent cytotoxic action against the liver and breast cancer cell line. Comparing compound 95 to doxorubicin (IC50 = 3.641 μg/mL), compound 95 is more effective against colon cancer cell lines (IC50 = 2.914 μg/mL).
2.5.4
Pyridine Derivatives Containing Acylhydrazone and Benzamide Moieties
Novel compound (E)-4-[(2-pyridylcarbonyl)diazanylidene)methyl]-N-(p-tolyl)benzamide (96) (Figure [30]) was tested against two human cancer cell lines and one human normal cell line to determine its antiproliferative properties.[89] Compound 96 exhibits remarkable antiproliferative efficacy against RPMI8226 cells with an IC50 of 0.12 ± 0.09 μM. Comparing compound 96 to imatinib, it was less hazardous. Compound 96 effectively inhibits cell growth as demonstrated by flow cytometry analysis, stopping the cell cycle at the G0 and G1 phases, which leads to the apoptosis of RPMI8226 cells by facilitating the release of mitochondrial ROS.
2.5.5
Pyridine Derivatives Containing a Pyrrole Moiety
The in vitro biological activities of pyridine derivatives containing a pyrrole moiety were assessed against maternal embryonic leucine zipper kinase (MELK). Compound 97 (Figure [31]) demonstrated a strong antiproliferative activity on MDA-MB-231, MCF-7, and A549 cell lines with IC50 = 0.109–0.245 μM.[90] showed outstanding enzyme inhibition at IC50 = 32 nM. Compound 97 effectively stops A549 cells in the G0/G1 phase and causes apoptosis in a dose-dependent manner according to flow cytometry studies.
2.5.6
3,5-Disubstituted 1H-Pyrazolo[3,4-b]pyridines as Multiacting Inhibitors

Against Microtubule and Kinases
Compound 98 showed excellent antiproliferative activities towards many cancer cell lines.[91] In both enzymatic and cellular experiments, compound 98 (Figure [31]) demonstrated a substantial inhibition of tubulin assembly and a potent inhibition towards FLT3 and Abl1. Compound 98 severely interfered with HUVEC tube formation and resulted in a cell cycle arrest at G2/M phase. Compound 98 at 10 mg/kg, suppressed tumor growth on the K562 leukemia xenograft model according to in vivo effectiveness tests.
2.6
Pyrrole Derivatives as Anticancer Agents
A prospective scaffold for antibacterial, antiviral, antimalarial, antitubercular, anticancer, and anti-inflammatory is the class of physiologically active heterocyclic molecules known as pyrrole derivatives.[92] By establishing hydrogen bonds with DNA, chemicals containing heteroatoms, like nitrogen, sulfur, and oxygen, strengthen the complex.[93] Typically, the anticancer activity of a chemical is correlated with the force of its contact with DNA. In addition, the intercalating chromophore has a polarized characteristic property and the best contact occurs when the chemical structure contains one or more nitrogen heteroatoms. Prodigiosin is well known anticancer drug containing pyrrole moiety.
In 2020, Raimondi and co-workers reviewed bioactive pyrrole-based compounds with target selectivity.[94]
2.6.1Pyrrole-Pyrimidine with Urea Derivatives
In 2019, Kilic-Kurt and co-workers[95] synthesized pyrrolo[2,3-d]pyrimidines with a urea moiety at C2 (Figure [32]) that show cytotoxic action against A549, PC-3, and MCF-7 cell lines. Cytotoxicity was impacted by the design of the scaffold. IC50 values for compounds 99, 100, and 101 of 0.35, 1.48, and 1.56 μM, respectively, showed that the A549 cells were very responsive to the therapy.

2.6.2
Phenylpyrroloquinolinone Derivatives
Carta et al.,[96] in 2017, examined the ability of new phenylpyrroloquinolinones (PPyQs) to inhibit the growth of a variety of solid tumor and leukemic cell lines (Figure [33]). Instead of having a sulfonyl or carbamoyl moiety at the pyrrole nitrogen, compound 102 has a benzoyl group. Compound 102 showed the best effect with GI50 = 0.2 nM against HeLa, 0.1 nM against HT-29, and 0.2 nM against MCF-7 cell lines while compound 103 which is a sulfonyl derivative was less potential as compared to compound 102. Compound 102 displayed good potential in blocking tubulin assembly (IC50 = 0.84 mM).

2.6.3
Pyrrolo[2,3-d]pyrimidines
Kurup et al.,[97] in 2018, reported the synthesis of eighteen pyrrolo[2,3-d]pyrimidines, dual inhibitors of both EGFR and Aurora kinase A (AURKA). These are involved in the control of several critical processes such as cell migration, differentiation, proliferation, and survival. All substances were tested for their potential to inhibit kinases using enzymatic assays. The findings demonstrated micromolar and nanomolar inhibition of AURKA and EGFR. Compound 104 (Figure [34]) showed excellent dual activity with IC50 of 1.99 mM against AURKA and 3.76 nM against EGFR.

2.6.4
Pyrrolo[3,2-c]pyridines
In 2018, El-Gamal and Oh reported the synthesis of a number of pyrrolo[3,2-c]pyridines and tested their inhibition of FMS kinase. With IC50 values of 60 and 30 nM, respectively compounds 105 and 106 (Figure [35]) demonstrated remarkable activity.[98] The antiproliferative properties of 106 were assessed against 13 cancer cell lines (breast, prostate, and ovarian), demonstrating selectivity for cancer cells over normal fibroblasts with an IC50 ranging from 0.15 to 1.78 μM. With an IC50 value of 84 nM, a strong effect was also seen against macrophages generated from bone marrow, blocking their CSF-1-induced proliferation.

2.6.5
Piperidine-3,4-diol and Piperidin-3-ol with Pyrrolotriazine Derivatives
Mesaros et al.,[99] in 2015, produced pyrrolo[2,1-f][1,2,4]triazine derivatives via diastereoselective synthesis and these compounds demonstrated good in vitro ALK inhibitory activity in both an enzyme assay (IC50 = 3–57 nM) and a cell-based assay (IC50 = 30–500 nM).
2.6.6
Pyrrolo[2,3-b]pyridines
In 2016, Kondapalli and co-workers[100] developed novel pyrrolo[2,3-b]pyridines and tested their antitumor efficaciousness against the human cancer cell lines MDA-MB-231, HeLa, and A549. Compounds 107a–k (Figure [36]) exhibited the greatest inhibition of growth at doses ranging from 0.17 to 25.9 μM.

2.6.7
Pyrrole Derivatives as CYP Inhibitors
A superfamily of enzymes known as cytochrome-P450 (CYP) includes members that are in charge of phase 1 metabolism.[94] Compound 108b (Figure [36]) preferentially inhibits CYP1B1 with IC50 = 0.21 μM while compound 108a inhibits all CYP1 isoforms with almost identical potency (IC50 = 0.9–1.1 μM).[101]
2.6.8
Pyrrolo[2,3-d]pyrimidin-2-amines
The in vitro cytotoxic activity of compound 109 (Figure [36]) was assessed against the human cancer cell lines HepG2 (liver), HCT116 (colorectal), and MCF-7 (breast). The epidermal growth factor receptor tyrosine kinase (EGFR-TK) inhibitory action of compound 109 was investigated and this compound displayed good inhibitory effect with IC50 = 0.107 μM.[80]
2.6.9
Pyrrole-Pyrimidine Derivatives as Multi-Kinase Inhibitors
The inhibitory effects of various compounds 110 (Figure [36]) on protein kinases were assessed.[102] Compound 110 (R = OMe) as compared to the reference drugs demonstrated good multi-kinase inhibitory activities in nanomolar range against EGFR, Her2, VEGFR2, and CDK2 protein kinases. Further evaluation of compound 110 revealed the ability to suppress cycle progression and induce programmed cell death.
2.7
Sulfonamides linked with heterocycles as Anticancer Agents
Belinostat, ABT-199, and amsacrine are anticancer drugs containing the sulfonamide moiety that have been approved by FDA.[118] Belinostat is a histone deacetylase inhibitor, ABT-199 is a Bc1-2 inhibitor, and amsacrine is a topoisomerase II inhibitor. The general structure of sulfonamides is of type RSO2NH2 where R may be an aliphatic or aromatic moiety, see, for example 111 (Figure [37]).[119] This structure has served as an essential fighting platform for a number of illnesses.
2.7.1Sulfonamide Benzoquinazolinones
The strong growth inhibitory effect of benzoquinazolines and sulfonamides against several cancer cells and TK enzymes motivated Soliman and co-workers to work on their molecular hybridization, which may result in a scaffold with promising anticancer activity (Figure [37]).[120] Compound 112 was used as reference in this study and anthraquinone derivative 113 showed excellent activity against HER2 with IC50 = 3.20 μM.
2.7.2
Metronidazole Acid Acyl and Phenylacetyl Benzenesulfonamide Derivatives
A family of N-acylsulfonamides of metronidazole was created by Zhu and co-workers as EGFR modulators.[121] Metronidazole belongs to the nitroimidazole family chemically. Protein and nucleic acid are readily damaged by its action which entails penetration and accumulation in the tumor region followed by bioreduction that produces electrophilic chemicals. The good biological activity and low toxicity of acyl sulfonamides prompted Zhu and co-workers to synthesize metronidazole acid sulfonamide derivatives 114 (Figure [37]). Using the same lengths of the phenylacetic acid and metronidazole side chains, a series of phenylacetyl benzene sulfonamides 115 was created. Compounds with a metronidazole skeleton had superior EGFR inhibitory activity (IC50 = 0.39–38.52 μM), whereas compounds containing phenylacetic acid have moderate EGFR inhibitory activity (IC50 = 5.17–38.52 μM). Excellent anticancer (IC50 = 1.26 μg/mL for A549 and IC50 = 0.35 μg/mL for B16-F10) and EGFR inhibition (IC50 = 0.39 μM for EGFR and 1.53 μM for HER-2) activities were demonstrated by molecule 116.


2.7.3
Quinazoline Sulfonamide Derivatives
Compounds 117 (Figure [38]) had remarkable cytotoxic activity against MCF-7, EGFR, and VEGFR with IC50 = 0.0977 μM, 0.0728 μM, and 0.0523 μM, respectively.[122] Compound 117 could bind to the ATP-binding site of EGF and VEGF receptors thereby decreasing their function as determined using molecular docking. The clinical significance of 4-anilinoquinazoline derivatives such as vandetanib, WHI-P180, gefitinib, lapatinib, afatinib, and erlotinib lies in their ability to decrease tumor growth by targeting either the VEGFR-2 or EGFR signaling pathways.
2.7.4
Sulfonamide-Pyrimidine Derivatives
Compound 118 (Figure [38]) showed excellent activity with IC50 = 0.0215 μM against H1975 cancer cell lines (EGFRT790M/L858R high express) and 0.011 μM against H2228 cells (ALK rearrangement).[123] The terminal amino group of the sulfonamide is a secondary amine with potent anticancer action. Its sulfonamide moiety has a cyclopropyl group attached to it. Compound 118 demonstrated strong kinase inhibitory activity with IC50 = 3.31 nM against EML4-ALK rearrangement and 17.74 nM against EGFRT790M/L858R mutation. The acrylamide moiety of 118 interacts with the cysteine residues in EGFR kinase, effectively inhibiting EGFRT790M/L858R. In order to assess the cytotoxicity of 118, two cell types were chosen: renal tubular cells (HK-2) and umbilical vein endothelial cells (EA.hy926). The MTT method was utilized for this purpose. Compound 118 has high selectivity for EA.hy926 normal cells with the selective ratio of 74.83 (IC50 of H1975 cells/IC50 of EA.hy926 cells = 1.609 μM/0.0215 μM) and 146.27 (1.609 μM/0.011 μM) to H1975 and H2228 cancer cell lines, respectively.
2.7.5
Diazepam-sulfonamides
Compound 119 (Figure [38]), which is a diazepam-bearing sulfonamide moiety, was evaluated for its anticancer activity against HepG2, HCT-116, and MCF-7 cell lines.[124] Compound 119 shown remarkable efficacy against cancer cell lines HepG2, HCT-116, and MCF-7 with IC50 = 8.98 ± 0.1 μM, 7.77 ± 0.1 μM, and 6.99 ± 0.1 μM, respectively. Compound 119 showed worse activity against the HCT116 cancer cell line but it displaced more activity than sorafenib against HepG2 and MCF-7. It showed superior activity against the HCT-116 cancer cell line but less activity against the HepG2 and MCF-7 cell lines when compared to doxorubicin. Compound 119 had remarkable efficacy against VEGFR-2, displaying IC50 = 0.10 ± 0.01 μM.
2.7.6
Chromone-Oxime Sulfonamide Derivatives
A chromone-oxime derivative containing a piperazine sulfonamide moiety was evaluated against indoleamine 2,3-dioxygenase 1 (IDO1).[125] Compound 120 (Figure [39]) demonstrated strong inhibitory activity with IC50 = 0.64 μM against hIDo1 and 1.04 μM against HeLa IDO1. A direct interaction between compound 120 and IDO1 protein was confirmed using surface plasmon resonance analysis. In MTT assay compound 120 displayed no cytotoxicity.

2.7.7
Tertiary Sulfonamide–Benzimidazole Derivatives
Compound 121, a tertiary sulfonamide derivative with a benzimidazole moiety (Figure [39]), showed good antiproliferative action against cell lines MGC-803, HGC-27, and SGC-7901 with IC50 = 1.02 μM, 1.61 μM, and 2.30 μM, respectively.[126] Compound 121 demonstrated strong cell-cancer and normal cell selectivity. Compound 121 was shown to interact with gastric cancer cell lines by disrupting the AKT/m-TOR and RAS/Raf/MEK/ERK pathways, as evidenced by the decrease in p-Akt and p-c-Raf.
2.7.8
Sulfonamide-Isomeric Triazole Hybrids
Aouad, Teleb, and co-workers synthesized a number of new sulfonamide-tethered isomeric triazole hybrids.[127] Of these, 122 (Figure [39]) was an excellent and the safest anticancer agent with IC50 values in nanomolar range of 7.37–11.96 nM. Compound 122 had IC50 = 5.66 nM against MMP-2 and 6.65 nM against VEGFR when compared to the reference MMP-2 inhibitor NNGH (IC50 = 299.50 nM) and the VEGFR-2 inhibitor sorafenib (IC50 = 4.92 nM). Compound 122 showed encouraging ligand efficiency metrics and in silico ADMET characteristics.
2.7.9
2-Phenylquinoline-4-carboxamides Bonded to Benzene
Sulfonamide containing quinolines were investigated for their inhibitory potential against four human (h) carbonic anhydrase (CA) isoforms hCA I, II, IX, and XII.[128] Compounds 123, 124, and 125 (Figure [40]) showed excellent inhibitory potential and their IC50 values in the nanomolar range are given in Table [3]; the standard drug chosen was acetazolamide (AAZ).

2.7.10
Carbazole Sulfonamide Derivatives
Compounds 126, 127, and 128 (Figure [41]) are N-substituted carbazole sulfonamide derivatives.[129] Compound 126 possesses excellent anticancer activity with IC50 = 19 nM against HepG2 whereas compound 127 and 128 also possess good anticancer activity with IC50 = 4.13 μM and 1.12 μM, respectively, against HepG2.
2.7.11
Cinnamic Acyl Sulfonamide Derivative
Compound 129 (Figure [41]), a cinnamic acyl sulfonamide derivative with a 1,3-benzodioxole group, is an excellent tubulin inhibitor with IC50 = 0.88 μM.[130] Docking simulations and 3D-QSAR of compound 129 were carried out.
2.8
Oxadiazole and Its Derivatives as Anticancer Compounds
Zibotentan, an oxadiazole based anti-cancer drug candidate developed by AstraZeneca is used for the treatment of prostate cancer.[131] Because it has unique pharmacokinetic properties and boosts the lipophilicity of the drug, 1,3,4-oxadiazole, one of the isomers of oxadiazole, has attracted the attention of researchers. The characteristics of this molecule help in the transmembrane diffusion of the medication to the target location.[132] Recently new topsentin-linked 1,3,4-oxadiazoles and indole-linked 1,3,4-oxadiazoles have demonstrated anticancer potential via tubulin polymerization inhibition.[133] [134]


1,3,4-Oxadiazoles Compounds Containing N-Heterocycles
Bhat et al. synthesized a series of 2-(N-heterocycle)-substituted 1,3,4-oxadiazoles.[135] Of these, 5-bromo-1-((4-chlorophenyl){[5-(4-hydroxyphenyl)-1,3,4-oxadiazol-2-yl]amino}methyl)indoline-2,3-dione (130, Figure [43]) displayed good cytotoxicity with IC50 = 0.78 μM against HT-29 and 0.26 μM against HepG2 by inhibiting EGFR and CDK2 kinases.
2.8.2
Amine Derivatives of 5-[5-(Chloromethyl)-1,3,4-oxadiazol-2-yl]-2-(4-fluorophenyl)pyridine
Vinayak et al. reported that {5-[6-(4-fluorophenyl)pyridin-3-yl]-1,3,4-oxadiazol-2-ylmethyl}phenylamine (131, Figure [43]) possesses cytotoxic activity with IC50 = 2.3 μM against Caco-2 cell lines.[136]

2.8.3
5-Pyridyl-1,3,4-oxadiazoles
Khalil and co-workers reported that N′-[(Z/E)-(3-indolyl)methylene]-2-{[5-(pyridin-4-yl)-1,3,4-oxadiazol-2-yl]sulfanyl}acetohydrazide (132, Figure [44]) possesses an anticancer effect with IC50 = 0.010 μM through inhibition of EGFR.[137]

2.8.4
1,3,4-Oxadiazole Derivatives as Tubulin Inhibitors
Compounds 133 and 134 (Figure [45]) shown superior sensitivity and good IC50 = 3.19–8.21 μM against colorectal HCT116, liver HepG2, and breast MCF-7 cancer cell lines when compared to colchicine.[138] Compounds 133 and 134 demonstrated exceptional IC50 = 7.95 and 9.81 nM, respectively, when evaluated for enzymatic activity against the tubulin enzyme.

2.8.5
1,2,3-Triazoles Containing 1,3,4-Oxadiazoles and 1,3,5-Triazines
1,2,3-Triazoles are nitrogen-containing five membered heterocyclic aromatic scaffolds with great significance in medicine. They demonstrated various biological activities, such as anticancer,[139] antimalarial,[140] antitubercular,[141] anti-HIV,[142] antifungal,[143] antibacterial,[144] antidiabetic,[145] [146] and antineoplastic.[147,148]
A new library of 1,2,3-triazole-incorporated 1,3,4-oxadiazole-triazine derivatives was designed, synthesized, and tested in vitro for anticancer activity against A549 (lung cancer), MCF-7 (breast cancer), and PC3 and DU-145 (prostate cancer) cell lines. The assessment employed the MIT assay with etoposide serving as the control medication.[149] With IC50 values ranging from 0.16 ± 0.083 μM to 11.8 ± 7.46 μM, the compounds demonstrated excellent anticancer efficacy. Compound 135 (Figure [45]) containing a 4-pyridyl moiety exhibits exceptional anticancer activity with IC50 = 0.17 ± 0.063 μM, 0.19 ± 0.075 μM, 0.51 ± 0.083 μM, and 0.16 ± 0.083 μM against PC3, A549, MCF-7, and DU-145 cell lines, respectively.
2.8.6
Oxadiazole Derivatives as HDAC Inhibitors
In 2022, Singh and co-workers reviewed oxadiazole derivatives as histone deacetylase inhibitors in anticancer therapy and drug discovery.[150]
In 2014, Mai and co-workers synthesized a series of 1,3,4-oxadiazole derivatives (Figure [46]) and these were evaluated on different types of HDAC enzymes.[151] (E)-N-(2-Aminophenyl)-3-(4-{[5-(naphthalen-1-ylmethyl)-1,3,4-oxadiazol-2-yl]methyl}phenyl)acrylamide (136) was found to be most potent and selective against HDAC1. Compound 136 exhibited remarkable antiproliferative properties against the five cancer cells associated with leukemia. Comparing compound 137, 138, and 139 to SAHA, they demonstrated superior HDAC1 inhibitory action. Hydroxamic acid derivatives 137 and 138 exhibited greater potency towards HDAC4/6 in comparison benzamide derivatives 136 and 139. Compound 136 displayed good activity with IC50 = 0.2 μM against HDAC1, 0.89 μM against HDAC6, 1.8 μM against U937, 2.8 μM against HL-60, 2 μM against HEL, 1.8 μM against KG1, and 1.6 μM against MOLM13, while compound 137 exhibits favorable IC50 = 0.2 μM for HDAC1, 0.03 μM for HDAC6, 2.6 μM for KG1, and 2.2 μM for MOLM13. Compound 138 had an IC50 = 0.2 μM against HDAC1 and 1.2 μM.


2.9
Benzothiazole-Triazole Hybrids as Anticancer Compounds
Benzothiazoles are an important class of heterocyclic compounds that have attracted great attention due to their antimicrobial,[152] antileishmanial,[153] antitumor,[154] and antiviral[155] properties. Many modified benzothiazole derivatives (Figure [47]) that inhibit topoisomerase II (compound 140),[156] β-glucoronidase (compound 141),[157] CYP1A1 of cytochrome P450 enzyme,[158] and tyrosine kinase histone deacetylase[159] enzymes have been reported. Compound 142 is a topo I inhibitor with IC50 = 8.2 μM. Compound 143 is a MARK4 inhibitor with IC50 = 8.4 μM. Compound 144 is a benzothiazole-triazole hybrid that possess anticancer activity, reported by Rawat et al.[160]
2.9.1Benzothiazole and Isatin Coupled to a 1,2,3-Triazole Moiety as EGFR Inhibitors
A number of medications including benzothiazole/isatin coupled to the 1,2,3-triazole moiety and sulfonamides were created and tested for their ability to kill a variety of cancer cell types.[161] Comparing compound 145 (Figure [48]) to erlotinib, it demonstrated better EGFR activity (IC50 = 103 vs 67.6 nM). Under HepG2 model testing, compound 145 demonstrated potent inhibition of tumor growth, a strong induction of cancer cell death and suppression of cell cycle progression leading to DNA fragmentation.

2.9.2
Apoptosis Inducing and Tubulin Polymerization-Inhibiting Compounds: Benzothiazole Incorporated with Triazole and Tetrazole Rings
The combretastatin pharmacophore was modified to synthesize a series of colchicine site binding tubulin inhibitors.[162] In order to limit the cis orientation of the olefinic bond, the triazole and tetrazole rings, which bore structural similarities to combretastatin, a tubulin inhibitor, were added. Compound 146 (Figure [48]) was the most effective molecule, exhibiting an antiproliferative action similar to CA-4 with an IC50 = 0.04 μM against the human lung cancer cell line (A549). Hoechst staining was used in compound 146 investigations to verify that the chemical caused apoptotic cell death.
2.9.3
Benzothiazole-Pyrimidine Derivatives
Diao et al. reported 2-aminobenzothiazole derivatives based on pyrimidines as strong anticancer agents. The selection process yielded the following five human anticancer cell lines: MCF-7, HeLa, PC-3, MDA-MB-231, and HCT116. The most effective molecule was discovered to be compound 147 which showed IC50 = 0.45, 0.70, 0.92, and 1.80 μM against HeLa, HCT116, PC-3, and MDA-MB-231, respectively.[163]
2.9.4
Benzothiazole-Amino Derivatives
Lee et al. reported the discovery of 2-aminobenzothiazole compounds that exhibit anticancer action and serve as Aurora B kinase inhibitors. A series of derivatives inhibitory activity against Aurora B kinase was assessed based on the activity of the enzyme at various concentrations. When evaluated at enzyme concentrations of 1 μM, compounds 148 and 149 (Figure [48]), which contain chlorine and bromine groups at the para-position of the phenyl ring, demonstrated excellent inhibitory activity against Aurora B kinase with IC50 = 0.12 and 0.09 μM.[164]
2.9.5
Amino-benzothiazole Urea Derivatives
Xie et al. reported the powerful anticancer, mTOR, and PI3K inhibitor properties of amino-benzothiazole urea derivatives. These compounds had a substituted pyridine ring at C6 of the 2-aminobenzothiazole. The HCT116, MCF-7, U87 MG, and A549 cancer cell lines were utilized to assess the anticancer efficacy of each derivative. In comparison to HCT116, MCF-7, U87 MG, and A549, compound 150 (Figure [49]) showed IC50 = 0.30 μM, 0.32 μM, 0.39 μM, and 0.45 μM, respectively.[165]

2.9.6
Morpholine-Substituted Benzothiazoles
Cao et al. demonstrated the use of benzothiazole derivatives as a powerful PI3K inhibitor and anticancer drug. The derivatives IC50 values against the cancer cell lines A549, MCF7, DU145, PC-3, and HepG2 were found to be between 0.35 and 10.93 μM. The results showed that 151 (Figure [49]) was the most effective against the human cancer cell lines PC-3, MCF7, DU145, HepG2, and A549 with IC50 = 0.35 μM, 0.36 μM, 0.62 μM, 3.43 μM, and 3.48 μM, respectively.[166]
2.9.7
Benzothiazole-Tertiary Amide Derivatives
Song et al. created tertiary amide compounds with a 2-mercaptobenzothiazole moiety that are powerful anticancer drugs. MGC803, HCT-116, and PC-3 are human cancer cell lines that were chosen because they showed good anticancer inhibitory potency with an IC50 value range of 0.035–16.4 μM. It was discovered that compounds 152 and 153 (Figure [49]) were highly effective against the HCT-116 cell line with IC50 = 0.491 μM, and 0.182 μM, respectively.[167]
2.9.8
Benzothiazole-Acylhydrazones
Osmaniye et al. reported a number of compounds of mercaptobenzothiazole acylhydrazones (Figure [50]) as effective anticancer drugs. They were tested on four human cancer cell lines: A549, C6, MCF-7, and HT-29 and had an IC50 value of less than 1.50 μM. Testing compound 154 against the cancer cell lines MCF-7, C6, and HT-29 it showed IC50 = 0.10 μM, 0.03 μM, and 0.30 μM, respectively. In contrast, compound 155 showed IC50 values against NIH3T3, A549, C6, and MCF-7 cell lines of 0.01 μM, 0.03 μM, 0.03 μM, and 0.30 μM, respectively.[168]

2.9.9
Benzothiazole-Pyrazoles
Belal and Abdelgawad created a number of unique benzothiazole-pyrazole hybrids as anticancer agents. Three distinct human cancer cell lines were chosen namely A549, Hep3B (hepatoma cells), and MCF-7. Compound 156 (Figure [50]) had remarkable inhibitory efficacy against the A549 cell line, demonstrating IC50 = 2.35 μM.[169]
2.9.10
Comberstatin-Benzothiazole Hybrids
Comberstatin-benzothiazole hybrids (Figure [50]) which are strong tubulin inhibitors, are structurally linked to the benzothiazole molecule by a 1,2,4-triazole bridge. Compound 157, with a methoxy group at C6 of the benzothiazole ring, showed strong inhibitory potency against the A549 cell line (IC50 = 0.054 μM) while against the HeLa, DU145, HepG2, and MCF-7 cancer cell line (IC50 = 0.16 μM, 0.48 μM, 0.67 μM, and 1.13 μM, respectively). Compound 158 the most potent of the series, was shown to exhibit remarkable anticancer activity against A549, HeLa, and DU-145, with IC50 = 0.048 μM, 0.15 μM, and 0.28 μM, respectively. Compound 158 was substituted with a fluorine at C6 of the benzothiazole moiety.[170]
2.9.11
Benzothiazole-Phthalimides
Benzothiazole-phthalimide hybrids as potent anticancer agents were reported by Philoppes and Lamie.[171] Compound 159 (Figure [50]) has IC50 = 0.098 μM, 28.82 μM, and 0.006 μM against the cancer cell lines HepG2, WI-38, and MCF-7, respectively.
3
Conclusion
Heterocyclic compounds are the most important organic compounds as they play an important role in preparation of drugs. A summary of biological activity of various heterocyclic anticancer compounds has been presented in this review. The major motivation for creating this review is to assist researchers in this field, as the abundance of studies conducted both historically and presently on the development of heterocyclic anticancer drugs underlines the importance of these molecules.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
The authors would like to thank Prof Diwan S Rawat for his guidance and Principal Gargi College for providing the infrastructure facility.
-
References
- 1 Arrousse N, Harras MF, El Kadiri S, Haldhar R, Ichou H, Bousta D, Grafov A, Rais Z, Taleb M. Results Chem. 2023; 6: 100996
- 2 Ioele G, Chieffallo M, Occhiuzzi MA, De Luca M, Garofalo A, Ragno G, Grande F. Molecules 2022; 27: 5436
- 3 Bray F, Laversanne M, Weiderpass E, Soerjomataram I. Cancer 2021; 127: 3029
- 4 Negi B, Kumar D, Rawat DS. Curr. Protein Pept. Sci. 2016; 18: 885
- 5 Malusare M, Papule P, Swami GM. Int. Res. J. Modernization Eng. Technol. Sci. 2023; 5: 3963
- 6 Crescioli S, Kaplon H, Chenoweth A, Wang L, Reichert JM, Crescioli S, Kaplon H, Chenoweth A, Wang L, Chenoweth A, Wang L. MAbs 2024; 16: 2297450
- 7 Gogia P, Ashraf H, Bhasin S, Xu Y. Cancers 2023; 15: 3886
- 8 Parashar RK, Negi B. Chemistry of Heterocyclic Compounds . Ane Books Pvt. Ltd./CRC Press; New Delhi/Boca Raton: 2015
- 9 Qadir T, Amin A, Sharma PK, Jeelani I, Abe H. Open Med. Chem. J. 2022; 16: e187410452209010
- 10 Chu X.-M, Wang C, Liu W, Liang L.-L, Gong K.-K, Zhao C.-Y, Sun K.-L. Eur. J. Med. Chem. 2019; 161: 101
- 11 Li S, Huang Q, Liu Y, Zhang X, Liu S, He C, Gong P. Eur. J. Med. Chem. 2013; 64: 62
- 12 Ilovaisky AI, Scherbakov AM, Merkulova VM, Chernoburova EI, Shchetinina MA, Andreeva OE, Salnikova DI, Zavarzin IV, Terent’ev AO. J. Steroid Biochem. Mol. Biol. 2023; 228: 106245
- 13 Amin MM, Abuo-Rahma GE.-D. A, Shaykoon MS. A, Marzouk AA, Abourehab MA. S, Saraya RE, Badr M, Sayed AM, Beshr EA. M. Bioorg. Chem. 2023; 134: 106444
- 14 Kardile RA, Sarkate AP, Lokwani DK, Tiwari SV, Azad R, Thopate SR. Eur. J. Med. Chem. 2023; 245: 114889
- 15 Jeleń M, Morak-Młodawska B, Korlacki R. J. Mol. Struct. 2023; 1287: 135700
- 16a Jain S, Chandra V, Kumar Jain P, Pathak K, Pathak D, Vaidya A. Arabian J. Chem. 2019; 12: 4920 ; and references cited therein
- 16b Ilakiyalakshmi M, Napoleon AA. Arabian J. Chem. 2022; 15: 104168
- 16c Kumar D, Negi B, Rawat DS. Recent Advances in Pharmaceutical Innovation and Research. Springer Nature; Singapore: 2023: 243-292
- 17 Jiang R, Duckett D, Chen W, Habel J, Ling YY, Grasso PL, Kamenecka TM. Bioorg. Med. Chem. Lett. 2007; 17: 6378
- 18 Bispo M. deL. F, de Alcantara CC, de Moraes MO, do Ó Pessoa C, Rodrigues FA. R, Kaiser CR, Wardell SM. S. V, Wardell JL, de Souza MV. N. Monatsh. Chem. 2015; 146: 2041
- 19 Ferrer R, Lobo G, Gamboa N, Rodrigues J, Abramjuk C, Jung K, Lein M, Charris JE. Sci. Pharm. 2009; 77: 725
- 20 Xiang PJ. H, Zhou Y, Yang B, Wang HJ, Hu J, Hu J, Yang SY, Zhao YL. Molecules 2015; 20: 7620
- 21 Jiang N, Zhai X, Li T, Liu D, Zhang T, Wang B, Gong P. Molecules 2012; 17: 5870
- 22 Chen YL, Huang CJ, Huang ZY, Tseng CH, Chang FS, Yang SH, Lin SR, Tzeng CC. Bioorg. Med. Chem. 2006; 14: 3098
- 23 Chen YL, Chung CH, Chen IL, Chen PH, Jeng HY. Bioorg. Med. Chem. 2002; 10: 2705
- 24 Işik A, Acar Çevik U, Çelik I, Bostancı HE, Karayel A, Gündoğdu G, Ince U, Koçak A, Özkay Y, Kaplancıklı ZA. J. Mol. Struct. 2022; 1270: 133946
- 25 Suárez-Moreno GV, Hernández-Romero D, García-Barradas Ó, Vázquez-Vera Ó, Rosete-Luna S, Cruz-Cruz CA, López-Monteon A, Carrillo-Ahumada J, Morales-Morales D, Colorado-Peralta R. Coord. Chem. Rev. 2022; 472: 214790
- 26 Banerjee S, Mukherjee S, Nath P, Mukherjee A, Mukherjee S, Kumar S K A, De S, Banerjee S. Results Chem. 2023; 6: 101013
- 27 Ebenezer O, Oyetunde-Joshua F, Omotoso OD, Shapi M. Results Chem. 2023; 5: 100925
- 28 Garuti L, Roberti M, Malagoli M, Rossi T, Castelli M. Bioorg. Med. Chem. Lett. 2000; 10: 2193
- 29 Chojnacki K, Winska P, Skierka K, Wielechowska M, Bretner M. Bioorg. Chem. 2017; 72: 1
- 30 Abd El-Meguid EA, El-Deen EM. M, Nael MA, Anwar MM. Arab. J. Chem. 2020; 13: 9179
- 31 Tang L, Wu W, Zhang C, Shi Z, Chen D, Zhai X, Jiang Y. Bioorg. Chem. 2021; 114: 105026
- 32 Eldehna WA, El Hassab MA, Abo-Ashour MF, Al-Warhi T, Elaasser MM, Safwat NA, Suliman H, Ahmed MF, Al-Rashood ST, Abdel-Aziz HA, El-Haggar R. Bioorg. Chem. 2021; 110: 104748
- 33 Nazreen S, Almalki AS. A, Elbehairi SE. I, Shati AA, Alfaifi MY, Elhenawy AA, Alsenani NI, Alfarsi A, Alhadhrami A, Alqurashi EA, Alam MM. Molecules 2022; 27: 6899
- 34 Lee YT, Tan YJ, Oon CE. Acta Pharm. Sin. B 2023; 13: 478
- 35 Karthikeyan C, Solomon VR, Lee H, Trivedi P. Arabian J. Chem. 2017; 10: S1788
- 36 Kamal A, Subba Rao AV. S, Lakshma Nayak VL, Subba Reddy NV. S, Swapna K, Ramakrishna G, Alvala M. Org. Biomol. Chem. 2014; 12: 9864
- 37 Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, Reece D, Heise C, Stewart AK. Blood 2005; 105: 2941
- 38 Roskoski R. Pharmacol. Res. 2019; 144: 19 ; and references cited therein
- 39 Corona SP, Generali D. Drug Des., Dev. Ther. 2018; 12: 321
- 40 Ramurthy S, Subramanian S, Aikawa M, Amiri P, Costales A, Dove J, Fong S, Jansen JM, Levine B, Ma S, McBride CM, Michaelian J, Pick T, Poon DJ, Girish S, Shafer CM, Stuart D, Sung L, Renhowe PA. J. Med. Chem. 2008; 51: 7049
- 41 Li Y, Tan C, Gao C, Zhang C, Luan X, Chen X, Liu H, Chen Y, Jiang Y. Bioorg. Med. Chem. 2011; 19: 4529
- 42 Galal SA, Khairat SH. M, Ali HI, Shouman SA, Attia YM, Ali MM, Mahmoud AE, Abdel-Halim AH, Fyiad AA, Tabll A, El-Shenawy R, El Abd YS, Ramdan R, El Diwani HI. Eur. J. Med. Chem. 2018; 144: 859
- 43 Bistrović A, Krstulović L, Harej A, Grbčić P, Sedić M, Koštrun S, Pavelić SK, Bajić M, Raić-Malić S. Eur. J. Med. Chem. 2018; 143: 1616
- 44 Zanjirband M, Curtin N, Edmondson RJ, Lunec J. Oncotarget 2017; 8: 69779
- 45 Akunuri R, Vadakattu M, Bujji S, Veerareddy V, Madhavi YV, Nanduri S. Eur. J. Med. Chem. 2021; 220: 113445
- 46 Konus M, Çetin D, Kızılkan ND, Yılmaz C, Fidan C, Algso M, Kavak E, Kivrak A, Kurt-Kızıldoğan A, Otur Ç, Mutlu D, Abdelsalam AH, Arslan S. J. Mol. Struct. 2022; 1263: 133168
- 47 Wan Y, Li Y, Yan C, Yan M, Tang Z. Eur. J. Med. Chem. 2019; 183: 111691
- 48 Nishiguchi GA, Atallah G, Bellamacina C, Burger MT, Ding Y, Feucht PH, Garcia PD, Han W, Klivansky L, Lindvall M. Bioorg. Med. Chem. Lett. 2011; 21: 6366
- 49a More KN, Jang HW, Hong VS, Lee J. Bioorg. Med. Chem. Lett. 2014; 24: 2424
- 49b More KN, Hong VS, Lee A, Park J, Kim S, Lee J. Bioorg. Med. Chem. Lett. 2018; 28: 2513
- 50 Dai Y, Guo Y, Guo J, Pease LJ, Li J, Marcotte PA, Glaser KB, Tapang P, Albert DH, Richardson PL, Davidsen SK, Michaelides MR. Bioorg. Med. Chem. Lett. 2003; 13: 1897
- 51 Giannini G, Marzi M, Di Marzo M, Battistuzzi G, Pezzi R, Brunetti T, Cabri W, Vesci L, Pisano C. Bioorg. Med. Chem. Lett. 2009; 19: 2840
- 52 Hawash M, Ergun SG, Kahraman DC, Olgac A, Hamel E, Cetin-Atalay R, Baytas SN. J. Mol. Struct. 2023; 1285: 135477
- 53 Li J, Si R, Zhang Q, Li Y, Zhang J, Shan Y. Chem.-Biol. Interact. 2022; 368: 110242
- 54 Wang G, He M, Liu W, Fan M, Li Y, Peng Z. Arabian J. Chem. 2022; 15: 103504
- 55 Wang G, He M, Liu W, Fan M, Li Y, Zhiyun Peng Z. Arabian Journal of Chem. 2022; 15: 103504
- 56 Peerzada MN, Khan P, Ahmad K, Hassan MI, Azam A. Eur. J. Med. Chem. 2018; 155: 13
- 57 Wang Q, Arnst KE, Wang Y, Kumar G, Ma D, White SW, Miller DD, Li W, Li W. J. Med. Chem. 2019; 62: 6734
- 58 He Z, Qiao H, Yang F, Zhou W, Gong Y, Zhang X, Wang H, Zhao B, Ma L, Liu H. Eur. J. Med. Chem. 2019; 184: 111764
- 59 El-Sharief AM. S, Ammar YA, Belal A, El-Sharief MA. M. S, Mohamed YA, Mehany AB. M, Ali GA. M. E, Ragab A. Bioorg. Chem. 2019; 85: 399
- 60 Iacopetta D, Catalano A, Ceramella J, Barbarossa A, Carocci A, Fazio A, La Torre C, Caruso A, Ponassi M, Rosano C. Bioorg. Chem. 2020; 105: 104440
- 61 Gurkan-Alp AS, Mumcuoglu M, Andac CA, Dayanc E, Cetin-Atalay R, Buyukbingol E. Eur. J. Med. Chem. 2012; 58: 346
- 62 Fortes MP, da Silva PB. N, da Silva TG, Kaufman TS, Militao GC. G, Silveira CC. Eur. J. Med. Chem. 2016; 118: 21
- 63 Ahmed SA, Naji TS, Mohammad FI. Al-Nahrain J. Sci. 2013; 16: 84
- 64 Mahapatra A, Prasad T, Sharma T. Future J. Pharm. Sci. 2021; 7: 123
- 65 Mahdy H, Elnagar H, Sakr H. Al-Azhar J. Pharm. Sci. 2023; 67: 51
- 66 Li Z, Kou L, Fu X, Xie Z, Xu M, Guo L, Lin T, Gong S, Zhang S, Liu M. J. Enzyme Inhib. Med. Chem. 2022; 37: 1514
- 67 Akhtar W, Nainwal LM, Khan MF, Verma G, Chashoo G, Bakht A, Iqbal M, Akhtar M, Shaquiquzzaman M, Alam MM. J. Fluorine Chem. 2020; 236: 109579
- 68 Haoran W, Akhtar W, Nainwal LM, Kaushik SK, Akhter M, Shaquiquzzaman M, Alam MM. J. Heterocycl. Chem. 2020; 57: 3350
- 69 Xu Y, Hao SY, Zhang XJ, Li WB, Qiao XP, Wang ZX, Chen SW. Bioorg. Med. Chem. Lett. 2020; 30: 126885
- 70 Hu G, Wang C, Xin X, Li S, Li Z, Zhao Y, Gong P. New J. Chem. 2019; 43: 10190
- 71 Qin WW, Sang CY, Zhang LL, Wei W, Tian HZ, Liu HX, Chen SW, Hui L. Eur. J. Med. Chem. 2015; 95: 174
- 72 Kraljević TG, Klika M, Kralj M, Martin-Kleiner I, Jurmanović S, Milić A, Padovan J, Raić-Malić S. Bioorg. Med. Chem. Lett. 2012; 22: 308
- 73 Weinberg LR, Albom MS, Angeles TS, Husten J, Lisko JG, McHugh RJ, Milkiewicz KL, Murthy S, Ott GR, Theroff JP, Tripathy R. Bioorg. Med. Chem. Lett. 2011; 21: 164
- 74 Long L, Luo Y, Hou ZJ, Ma HJ, Long ZJ, Tu ZC, Huang LJ, Liu Q, Lu G. Eur. J. Med. Chem. 2018; 145: 805
- 75 Luo G, Tang Z, Lao K, Li X, You Q, Xiang H. Eur. J. Med. Chem. 2018; 150: 783
- 76 Zhao M, Ren H, Chang J, Zhang D, Yang Y, He Y, Qi C, Zhang H. Eur. J. Med. Chem. 2016; 119: 183
- 77 Jorda R, Havlíček L, Šturc A, Tušková D, Daumová L, Alam M, Škerlová J, Nekardová M, Peřina M, PospíšilT ŠJ. J. Med. Chem. 2019; 62: 4606
- 78 Jorda R, Havlícek L, McNae IW, Walkinshaw MD, Voller J, Šturc A, Navrátilová J, Kuzma M, Mistrík M, Bártek J, Strnad M. J. Med. Chem. 2011; 54: 2980
- 79 Wang R, Chen Y, Zhao X, Yu S, Yang B, Wu T, Guo J, Hao C, Zhao D, Cheng M. Eur. J. Med. Chem. 2019; 183: 111716
- 80 Sivaiah G, Raveesha R, Benaka Prasad SB, Yogesh Kumar K, Raghu MS, Alharti FA, Prashanth MK, Jeon B.-H. J. Mol. Struct. 2023; 1275: 134728
- 81 Alrooqi M, Khan S, Alhumaydhi FA, Asiri SA, Alshamrani M, Mashraqi MM, Alzamami A, Alshahrani AM, Aldahish AA. Anti-Cancer Agents Med. Chem. 2022; 22: 2775
- 82 Marinescu M, Popa CV. Int. J. Mol. Sci. 2022; 23: 5659
- 83 De S, Kumar S K A, Kumar Shah S, Kazi S, Sarkar N, Banerjee S, Deya S. RSC Adv. 2022; 12: 15385
- 84 Albratty M, Alhazmi HA. Arabian J. Chem. 2022; 15: 103846
- 85 Narasimhamurthy KH, Kallesha N, Mohan CD, Rangappa KS. Anticancer Functions of Pyridine Heterocycles . In Cytotoxicity - Understanding Cellular Damage and Response . Sukumaran A, Mansour MA. IntechOpen; London: 2023
- 86 El-Naggar M, Almahli H, Ibrahim HS, Eldehna WM, Abdel-Aziz HA. Molecules 2018; 23: 1459
- 87 Murthy IS, Sreenivasulu R, Alluraiah G, Ramesh Raju G. Russ .J. Gen. Chem. 2019; 89(8): 1718
- 88 Metwally NH, Deeb EA. Bioorg. Chem. 2018; 77: 203
- 89 Li X, Li S, Lu G, Wang D, Liu K, Qian X, Xue W, Meng F. Bioorg. Chem. 2020; 103: 104189
- 90 Wang R, Chen Y, Yang B, Yu S, Zhao X, Zhang C, Hao C, Zhao D, Cheng M. Bioorg. Chem. 2020; 94: 103474
- 91 Ning C, Tao A, Xu J. Eur. J. Med. Chem. 2023; 259: 115687
- 92 Gholap SS. Eur. J. Med. Chem. 2016; 110: 13
- 93 Akhtar J, Khan AA, Ali Z, Haider R, Shahar Yar M. Eur. J. Med. Chem. 2017; 125: 143
- 94 Petri GL, Spanò V, Spatola R, Holl R, Raimondi MV, Barraja P, Montalbano A. Eur. J. Med. Chem. 2020; 208: 112783
- 95 Kilic-Kurt Z, Bakar-Ates F, Aka Y, Kutuk O. Bioorg. Chem. 2019; 83: 511
- 96 Carta D, Bortolozzi R, Sturlese M, Salmaso V, Hamel E, Basso G, Calderan L, Quintieri L, Moro S, Viola G, Ferlin MG. Eur. J. Med. Chem. 2017; 127: 643
- 97 Kurup S, McAllister B, Liskova P, Mistry T, Fanizza A, Stanford D, Slawska J, Keller U, Hoellein A. J. Enzyme Inhibit. Med. Chem. 2018; 33: 74
- 98 El-Gamal MI, Oh CH. J. Enzyme Inhibit. Med. Chem. 2018; 33: 1160
- 99 Mesaros EF, Angeles TS, Albom MS, Wagner JC, Aimone LD, Wan W, Lu L, Huang Z, Olsen M, Kordwitz E, Haltiwanger RC, Landis AJ, Cheng M, Ruggeri BA, Ator MA, Dorsey BD, Ott GR. Bioorg. Med. Chem. Lett. 2015; 25: 1047
- 100 Narva S, Chitti S, Bala BR, Alvala M, Jain N, Kondapalli VG. C. S. Eur. J. Med. Chem. 2016; 114: 220
- 101 Williams IS, Joshi P, Gatchie L, Sharma M, Satti NK, Vishwakarma RA, Chaudhuri B, Bharate SB. Bioorg. Med. Chem. Lett. 2017; 27: 3683
- 102 Alanazi AS, Mirgany TO, Alsaif NA, Alsfouk AA, Alanazi MM. Saudi Pharm. J. 2023; 31: 989
- 103 Niedziałkowski P, Czaczyk E, Jarosz J, Wcisło A, Białobrzeska W, Wietrzyk J, Ossowski T. J. Mol. Struct. 2019; 1175: 488
- 104 Tikhomirov AS, Shtil AA, Shchekotikhin AE. Recent Pat. Anti-Cancer Drug Discovery 2018; 13: 159
- 105 Anifowose A, Agbowuro AA, Tripathi R, Lu W, Tan C, Yang X, Wang B. Med. Chem. Res. 2020; 29: 1199
- 106 Zheng Y, Zhu L, Fan L, Zhao W, Wang J, Hao X, Zhu Y, Hu X, Yuan Y, Shao J, Wang W. Eur. J. Med. Chem. 2017; 125: 902
- 107 Choudhary N, Collignon TE, Tewari D, Bishayee A. Phytomedicine 2022; 105: 154356
- 108 Dobson J, de Queiroz GF, Golding JP. Vet. J. 2018; 233: 8
- 109 Kwiatkowski S, Knap B, Przystupski D, Saczko J, Kędzierska E, Knap-Czop K, Kotlińska J, Michel O, Kotowski K, Kulbacka J. Biomed. Pharmacother. 2018; 106: 1098
- 110 Champeau M, Vignoud S, Mortier L, Mordon S. J. Photochem. Photobiol., B 2019; 197: 111544
- 111 Woźniak M, Nowak M, Lazebna A, Więcek K, Jabłońska I, Szpadel K, Grzeszczak A, Gubernator J, Ziółkowski P. Pharmaceuticals 2021; 14: 374
- 112 Banerjee SM, El-Sheikh S, Malhotra A, Mosse CA, Parker S, Williams NR, Macrobert AJ, Hamoudi R, Bown SG, Keshtgar MR. S. J. Clin. Med. 2020; 9: 483
- 113 Ostańska E, Aebisher D, Bartusik-Aebisher D. Biomed. Pharmacother. 2021; 137: 111302
- 114 Afanasiev MS, Dushkin AD, Grishacheva TG, Afanasiev SS, Karaulov AV. Photodiagn. Photodyn. Ther. 2022; 37: 102620
- 115 Warowicka A, Popenda Ł, Bartkowiak G, Musidlak O, Litowczenko-Cybulska J, Kuźma D, Nawrot R, Jurga S, Goździcka-Józefiak A. Phytomedicine 2019; 64: 152919
- 116 Nowak-Perlak M, Ziółkowski P, Woźniak M. Phytomedicine 2023; 119: 155035
- 117 Stanojković T, Marković V, Matić IZ, Mladenović MP, Petrović N, Krivokuća A, Petković M, Joksović MD. Bioorg. Med. Chem. Lett. 2018; 28: 2593
- 118 Wan Y, Fang G, Chen H, Deng X, Tang Z. Eur. J. Med. Chem. 2021; 226: 113837
- 119 Das R, Tambe G, Shard A. Results Chem. 2023; 5: 100950
- 120 Soliman AM, Alqahtani AS, Ghorab MM. J. Enzyme Inhibit. Med. Chem. 2019; 34: 1030
- 121 Luo Y, Li Y, Qiu K.-M, Lu X, Fu J, Zhu H.-L. Bioorg. Med. Chem. 2011; 19: 6069
- 122 Ghorab MM, Soliman AM, El-Adl K, Hanafy NS. Bioorg. Chem. 2023; 140: 106791
- 123 Cao L, Yao H, Yu L, Ren Y, Liu J, Li X, Jia X. Bioorg. Med. Chem. 2023; 85: 117241
- 124 Saleh NM, El-Gaby MS. A, El-Adl K, Abd El-Sattar NE. A. Bioorg. Chem. 2020; 104: 104350
- 125 Wang K, Song L.-H, Liang Q.-L, Zhang Y, Ma X.-L, Wang Q, Zhang H.-Y, Jiang C.-N, Wei J.-H, Huang R.-Z. Eur. J. Med. Chem. 2023; 254: 115349
- 126 Song J, Gao Q.-L, Wu B.-W, Li D, Shi L, Zhu T, Lou J.-F, Jin C.-Y, Zhang Y.-B, Zhang S.-Y, Liu H.-M. Eur. J. Med. Chem. 2019; 183: 111731
- 127 Reda Aouad M, Almehmadi MA, Faleh Albelwi F, Teleb M, Tageldin GN, Abu-Serie MM, Hagar M, Rezki N. Bioorg. Chem. 2022; 124: 105816
- 128 Swain B, Sahoo SK, Singh P, Angeli A, Yaddanapudi VM, Supuran CT, Arifuddin M. Eur. J. Med. Chem. 2022; 234: 114247
- 129 Liu Y, Wu Y, Sun L, Gu Y, Hu L. Eur. J. Med. Chem. 2020; 191: 112181
- 130 Luo Y, Zhou Y, Song Y, Chen G, Wang Y.-X, Tian Y, Fan W.-W, Yang Y.-S, Cheng T, Zhu H.-L. Bioorg. Med. Chem. Lett. 2018; 28: 3634
- 131 For a review see: Nayak S, Gaonkar SL, Musad EA, Dawsar AM. A. L. J. Saudi Chem. Soc. 2021; 25: 101284
- 132 Boström J, Hogner A, Llinàs A, Wellner E, Plowright AT. J. Med. Chem. 2012; 55: 1817
- 133 Naaz F, Ahmad F, Lone BA, Pokharel YR, Fuloria NK, Fuloria S, Ravichandran M, Pattabhiraman L, Shafi S, Shahar Yar M. Bioorg. Chem. 2020; 95: 103519
- 134 Tantak MP, Malik M, Klingler L, Olson Z, Kumar A, Sadana R, Kumar D. Bioorg. Med. Chem. Lett. 2021; 37: 127842
- 135 Bhatt P, Sen A, Jha A. ChemistrySelect 2020; 5: 3347
- 136 Vinayak A, Sudha M, Lalita KS. Dhaka Univ. J. Pharm. Sci. 2017; 16: 11
- 137 Khalil NA, Kamal AM, Emam SH. Biol. Pharm. Bull. 2015; 38: 763
- 138 Yousef TA, Alhamzani AG, Abou-Krisha MM, Kanthimathi G, Raghu MS, Kumar KY, Prashanth MK, Jeon BH. Heliyon 2023; 9: e20300
- 139 Cho S, Oh S, Um Y, Jung J.-H, Ham J, Shin W.-S, Lee S. Bioorg. Med. Chem. Lett. 2009; 19: 382
- 140 Kumar K, Pradines B, Madamet M, Amalvict R, Kumar V. Eur. J. Med. Chem. 2014; 86: 113
- 141 Karthik Kumar K, Prabu Seenivasan S, Kumar V, Mohan Das T. Carbohydr. Res. 2011; 346: 2084
- 142 de Carvalho da Silva F, de Souza MC. B. V, Frugulhetti II. P, Castro HC, Souza SL. de O, de Souza TM. L, Rodrigues DQ, Souza AM. T, Abreu PA, Passamani F, Rodrigues CR, Ferreira VF. Eur. J. Med. Chem. 2009; 44: 373
- 143 Costa MS, Boechat N, Rangel ÉA, de Carvalho da Silva F, de Souza AM. T, Rodrigues CR, Castro HC, Junior IN, Lourenço MC. S, Wardell SM. S. V, Ferreira VF. Bioorg. Med. Chem. 2006; 14: 8644
- 144 Lal K, Kaushik CP, Kumar A. Med. Chem. Res. 2015; 24: 3258
- 145 Bokor É, Docsa T, Gergely P, Somsák L. Bioorg. Med. Chem. 2010; 18: 1171
- 146 Fallah Z, Tajbakhsh M, Alikhani M, Larijani B, Faramarzi MA, Hamedifar H, Mohammadi-Khanaposhtani M, Mahdavi M. J. Mol. Struct. 2022; 1255: 132469
- 147 Al-Masoudi NA, Al-Soud YA. Tetrahedron Lett. 2002; 43: 4021
- 148 Shawish I, Barakat A, Aldalbahi A, Alshaer W, Daoud F, Alqudah DA, Al Zoubi M, Hatmal MM, Nafie MS, Haukka M, Sharma A, de la Torre BG, Albericio F, El-Faham A. Pharmaceutics 2022; 14: 145
- 149 Oggu S, Akshinthala P, Katari NK, Nagarapu LK, Malempati S, Gundla R, Jonnalagadda SB. Heliyon 2023; 9: e15935
- 150 Matore BW, Banjare P, Guria T, Roy PP, Singh J. Eur. J. Med. Chem. Rep. 2022; 5: 100058
- 151 Valente S, Trisciuoglio D, De Luca T, Nebbioso A, Labella D, Lenoci A, Bigogno C, Dondio G, Miceli M, Brosch G, Del Bufalo D, Altucci L, Mai A. J. Med. Chem. 2014; 57: 6259
- 152 Bondock S, Fadaly W, Metwally MA. Eur. J. Med. Chem. 2010; 45: 3692
- 153 Delmas F, Avellaneda A, Di Giorgio C, Robin M, De Clercq E, Timon-David P, Galy J.-P. Eur. J. Med. Chem. 2004; 39: 685
- 154 Hong S, Kim J, Yun S.-M, Lee H, Park Y, Hong S.-S, Hong S. J. Med. Chem. 2013; 56: 3531
- 155 Ke S, Wei Y, Yang Z, Wang K, Liang Y, Shi L. Bioorg. Med. Chem. Lett. 2013; 23: 5131
- 156 Kaplan-Ozen C, Tekiner-Gulbas B, Foto E, Yildiz I, Diril N, Aki E, Yalcin I. Med. Chem. Res. 2013; 22: 5798
- 157 Khan KM, Rahim F, Wadood A, Taha M, Khan M, Naureen S, Ambreen N, Hussain S, Perveen S, Choudhary MI. Bioorg. Med. Chem. Lett. 2014; 24: 1825
- 158 O’Brien SE, Browne HL, Bradshaw TD, Westwell AD, Stevens MF. G, Laughton CA. Org. Biomol. Chem. 2003; 1: 493
- 159 Oanh DT. K, Van Hai H, Park SH, Kim HJ, Han BW, Kim HS, Hong JT, Han SB, Nam NH. Bioorg. Med. Chem. Lett. 2011; 21: 7509
- 160 Rawat S, Rawat DS, Negi B. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2021; 60: 409
- 161 Rezki N, Almehmadi MA, Ihmaid S, Shehata AM, Omar AM, Ahmed HE. A, Aouad MR. Bioorg. Chem. 2020; 103: 104133
- 162 Subba Rao AV, Swapna K, Shaik SP, Lakshma Nayak V, Srinivasa Reddy T, Sunkari S, Shaik TB, Bagul C, Kamal A. Bioorg. Med. Chem. 2017; 25: 977
- 163 Diao PC, Lin WY, Jian XE, Li YH, You WW, Zhao PL. Eur. J. Med. Chem. 2019; 179: 196
- 164 Lee E, An Y, Kwon J, Kim KI, Jeon R. Bioorg. Med. Chem. 2017; 25: 3614
- 165 Xie X.-X, Li H, Wang J, Mao S, Xin M.-H, Lu S.-M, Mei Q.-B, Zhang S.-Q. Bioorg. Med. Chem. 2015; 23: 6477
- 166 Cao S, Cao R, Liu X, Luo X, Zhong W. Molecules 2016; 21: 6
- 167 Song J, Gao QL, Wu BW, Zhu T, Cui XX, Jin CJ, Wang SY, Wang SH, Fu DJ, Liu HM, Zhang SY, Zhang YB, Li YC. Eur. J. Med. Chem. 2020; 203: 112618
- 168 Osmaniye D, Levent S, Karaduman AB, Ilgın S, Zkay Y, Kaplancikli ZA. Molecules 2018; 23: 2033
- 169 Belal A, Abdelgawad MA. Res. Chem. Intermed. 2017; 43: 3859
- 170 Haider K, Shrivastava N, Pathak A, Prasad Dewangan R, Yahya S, Shahar Yar M. Results Chem. 2022; 4: 100258
- 171 Philoppes JN, Lamie PF. Bioorg. Chem. 2019; 89: 102978
Corresponding Author
Publication History
Received: 13 March 2024
Accepted after revision: 12 June 2024
Article published online:
26 August 2024
© 2024. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Arrousse N, Harras MF, El Kadiri S, Haldhar R, Ichou H, Bousta D, Grafov A, Rais Z, Taleb M. Results Chem. 2023; 6: 100996
- 2 Ioele G, Chieffallo M, Occhiuzzi MA, De Luca M, Garofalo A, Ragno G, Grande F. Molecules 2022; 27: 5436
- 3 Bray F, Laversanne M, Weiderpass E, Soerjomataram I. Cancer 2021; 127: 3029
- 4 Negi B, Kumar D, Rawat DS. Curr. Protein Pept. Sci. 2016; 18: 885
- 5 Malusare M, Papule P, Swami GM. Int. Res. J. Modernization Eng. Technol. Sci. 2023; 5: 3963
- 6 Crescioli S, Kaplon H, Chenoweth A, Wang L, Reichert JM, Crescioli S, Kaplon H, Chenoweth A, Wang L, Chenoweth A, Wang L. MAbs 2024; 16: 2297450
- 7 Gogia P, Ashraf H, Bhasin S, Xu Y. Cancers 2023; 15: 3886
- 8 Parashar RK, Negi B. Chemistry of Heterocyclic Compounds . Ane Books Pvt. Ltd./CRC Press; New Delhi/Boca Raton: 2015
- 9 Qadir T, Amin A, Sharma PK, Jeelani I, Abe H. Open Med. Chem. J. 2022; 16: e187410452209010
- 10 Chu X.-M, Wang C, Liu W, Liang L.-L, Gong K.-K, Zhao C.-Y, Sun K.-L. Eur. J. Med. Chem. 2019; 161: 101
- 11 Li S, Huang Q, Liu Y, Zhang X, Liu S, He C, Gong P. Eur. J. Med. Chem. 2013; 64: 62
- 12 Ilovaisky AI, Scherbakov AM, Merkulova VM, Chernoburova EI, Shchetinina MA, Andreeva OE, Salnikova DI, Zavarzin IV, Terent’ev AO. J. Steroid Biochem. Mol. Biol. 2023; 228: 106245
- 13 Amin MM, Abuo-Rahma GE.-D. A, Shaykoon MS. A, Marzouk AA, Abourehab MA. S, Saraya RE, Badr M, Sayed AM, Beshr EA. M. Bioorg. Chem. 2023; 134: 106444
- 14 Kardile RA, Sarkate AP, Lokwani DK, Tiwari SV, Azad R, Thopate SR. Eur. J. Med. Chem. 2023; 245: 114889
- 15 Jeleń M, Morak-Młodawska B, Korlacki R. J. Mol. Struct. 2023; 1287: 135700
- 16a Jain S, Chandra V, Kumar Jain P, Pathak K, Pathak D, Vaidya A. Arabian J. Chem. 2019; 12: 4920 ; and references cited therein
- 16b Ilakiyalakshmi M, Napoleon AA. Arabian J. Chem. 2022; 15: 104168
- 16c Kumar D, Negi B, Rawat DS. Recent Advances in Pharmaceutical Innovation and Research. Springer Nature; Singapore: 2023: 243-292
- 17 Jiang R, Duckett D, Chen W, Habel J, Ling YY, Grasso PL, Kamenecka TM. Bioorg. Med. Chem. Lett. 2007; 17: 6378
- 18 Bispo M. deL. F, de Alcantara CC, de Moraes MO, do Ó Pessoa C, Rodrigues FA. R, Kaiser CR, Wardell SM. S. V, Wardell JL, de Souza MV. N. Monatsh. Chem. 2015; 146: 2041
- 19 Ferrer R, Lobo G, Gamboa N, Rodrigues J, Abramjuk C, Jung K, Lein M, Charris JE. Sci. Pharm. 2009; 77: 725
- 20 Xiang PJ. H, Zhou Y, Yang B, Wang HJ, Hu J, Hu J, Yang SY, Zhao YL. Molecules 2015; 20: 7620
- 21 Jiang N, Zhai X, Li T, Liu D, Zhang T, Wang B, Gong P. Molecules 2012; 17: 5870
- 22 Chen YL, Huang CJ, Huang ZY, Tseng CH, Chang FS, Yang SH, Lin SR, Tzeng CC. Bioorg. Med. Chem. 2006; 14: 3098
- 23 Chen YL, Chung CH, Chen IL, Chen PH, Jeng HY. Bioorg. Med. Chem. 2002; 10: 2705
- 24 Işik A, Acar Çevik U, Çelik I, Bostancı HE, Karayel A, Gündoğdu G, Ince U, Koçak A, Özkay Y, Kaplancıklı ZA. J. Mol. Struct. 2022; 1270: 133946
- 25 Suárez-Moreno GV, Hernández-Romero D, García-Barradas Ó, Vázquez-Vera Ó, Rosete-Luna S, Cruz-Cruz CA, López-Monteon A, Carrillo-Ahumada J, Morales-Morales D, Colorado-Peralta R. Coord. Chem. Rev. 2022; 472: 214790
- 26 Banerjee S, Mukherjee S, Nath P, Mukherjee A, Mukherjee S, Kumar S K A, De S, Banerjee S. Results Chem. 2023; 6: 101013
- 27 Ebenezer O, Oyetunde-Joshua F, Omotoso OD, Shapi M. Results Chem. 2023; 5: 100925
- 28 Garuti L, Roberti M, Malagoli M, Rossi T, Castelli M. Bioorg. Med. Chem. Lett. 2000; 10: 2193
- 29 Chojnacki K, Winska P, Skierka K, Wielechowska M, Bretner M. Bioorg. Chem. 2017; 72: 1
- 30 Abd El-Meguid EA, El-Deen EM. M, Nael MA, Anwar MM. Arab. J. Chem. 2020; 13: 9179
- 31 Tang L, Wu W, Zhang C, Shi Z, Chen D, Zhai X, Jiang Y. Bioorg. Chem. 2021; 114: 105026
- 32 Eldehna WA, El Hassab MA, Abo-Ashour MF, Al-Warhi T, Elaasser MM, Safwat NA, Suliman H, Ahmed MF, Al-Rashood ST, Abdel-Aziz HA, El-Haggar R. Bioorg. Chem. 2021; 110: 104748
- 33 Nazreen S, Almalki AS. A, Elbehairi SE. I, Shati AA, Alfaifi MY, Elhenawy AA, Alsenani NI, Alfarsi A, Alhadhrami A, Alqurashi EA, Alam MM. Molecules 2022; 27: 6899
- 34 Lee YT, Tan YJ, Oon CE. Acta Pharm. Sin. B 2023; 13: 478
- 35 Karthikeyan C, Solomon VR, Lee H, Trivedi P. Arabian J. Chem. 2017; 10: S1788
- 36 Kamal A, Subba Rao AV. S, Lakshma Nayak VL, Subba Reddy NV. S, Swapna K, Ramakrishna G, Alvala M. Org. Biomol. Chem. 2014; 12: 9864
- 37 Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, Reece D, Heise C, Stewart AK. Blood 2005; 105: 2941
- 38 Roskoski R. Pharmacol. Res. 2019; 144: 19 ; and references cited therein
- 39 Corona SP, Generali D. Drug Des., Dev. Ther. 2018; 12: 321
- 40 Ramurthy S, Subramanian S, Aikawa M, Amiri P, Costales A, Dove J, Fong S, Jansen JM, Levine B, Ma S, McBride CM, Michaelian J, Pick T, Poon DJ, Girish S, Shafer CM, Stuart D, Sung L, Renhowe PA. J. Med. Chem. 2008; 51: 7049
- 41 Li Y, Tan C, Gao C, Zhang C, Luan X, Chen X, Liu H, Chen Y, Jiang Y. Bioorg. Med. Chem. 2011; 19: 4529
- 42 Galal SA, Khairat SH. M, Ali HI, Shouman SA, Attia YM, Ali MM, Mahmoud AE, Abdel-Halim AH, Fyiad AA, Tabll A, El-Shenawy R, El Abd YS, Ramdan R, El Diwani HI. Eur. J. Med. Chem. 2018; 144: 859
- 43 Bistrović A, Krstulović L, Harej A, Grbčić P, Sedić M, Koštrun S, Pavelić SK, Bajić M, Raić-Malić S. Eur. J. Med. Chem. 2018; 143: 1616
- 44 Zanjirband M, Curtin N, Edmondson RJ, Lunec J. Oncotarget 2017; 8: 69779
- 45 Akunuri R, Vadakattu M, Bujji S, Veerareddy V, Madhavi YV, Nanduri S. Eur. J. Med. Chem. 2021; 220: 113445
- 46 Konus M, Çetin D, Kızılkan ND, Yılmaz C, Fidan C, Algso M, Kavak E, Kivrak A, Kurt-Kızıldoğan A, Otur Ç, Mutlu D, Abdelsalam AH, Arslan S. J. Mol. Struct. 2022; 1263: 133168
- 47 Wan Y, Li Y, Yan C, Yan M, Tang Z. Eur. J. Med. Chem. 2019; 183: 111691
- 48 Nishiguchi GA, Atallah G, Bellamacina C, Burger MT, Ding Y, Feucht PH, Garcia PD, Han W, Klivansky L, Lindvall M. Bioorg. Med. Chem. Lett. 2011; 21: 6366
- 49a More KN, Jang HW, Hong VS, Lee J. Bioorg. Med. Chem. Lett. 2014; 24: 2424
- 49b More KN, Hong VS, Lee A, Park J, Kim S, Lee J. Bioorg. Med. Chem. Lett. 2018; 28: 2513
- 50 Dai Y, Guo Y, Guo J, Pease LJ, Li J, Marcotte PA, Glaser KB, Tapang P, Albert DH, Richardson PL, Davidsen SK, Michaelides MR. Bioorg. Med. Chem. Lett. 2003; 13: 1897
- 51 Giannini G, Marzi M, Di Marzo M, Battistuzzi G, Pezzi R, Brunetti T, Cabri W, Vesci L, Pisano C. Bioorg. Med. Chem. Lett. 2009; 19: 2840
- 52 Hawash M, Ergun SG, Kahraman DC, Olgac A, Hamel E, Cetin-Atalay R, Baytas SN. J. Mol. Struct. 2023; 1285: 135477
- 53 Li J, Si R, Zhang Q, Li Y, Zhang J, Shan Y. Chem.-Biol. Interact. 2022; 368: 110242
- 54 Wang G, He M, Liu W, Fan M, Li Y, Peng Z. Arabian J. Chem. 2022; 15: 103504
- 55 Wang G, He M, Liu W, Fan M, Li Y, Zhiyun Peng Z. Arabian Journal of Chem. 2022; 15: 103504
- 56 Peerzada MN, Khan P, Ahmad K, Hassan MI, Azam A. Eur. J. Med. Chem. 2018; 155: 13
- 57 Wang Q, Arnst KE, Wang Y, Kumar G, Ma D, White SW, Miller DD, Li W, Li W. J. Med. Chem. 2019; 62: 6734
- 58 He Z, Qiao H, Yang F, Zhou W, Gong Y, Zhang X, Wang H, Zhao B, Ma L, Liu H. Eur. J. Med. Chem. 2019; 184: 111764
- 59 El-Sharief AM. S, Ammar YA, Belal A, El-Sharief MA. M. S, Mohamed YA, Mehany AB. M, Ali GA. M. E, Ragab A. Bioorg. Chem. 2019; 85: 399
- 60 Iacopetta D, Catalano A, Ceramella J, Barbarossa A, Carocci A, Fazio A, La Torre C, Caruso A, Ponassi M, Rosano C. Bioorg. Chem. 2020; 105: 104440
- 61 Gurkan-Alp AS, Mumcuoglu M, Andac CA, Dayanc E, Cetin-Atalay R, Buyukbingol E. Eur. J. Med. Chem. 2012; 58: 346
- 62 Fortes MP, da Silva PB. N, da Silva TG, Kaufman TS, Militao GC. G, Silveira CC. Eur. J. Med. Chem. 2016; 118: 21
- 63 Ahmed SA, Naji TS, Mohammad FI. Al-Nahrain J. Sci. 2013; 16: 84
- 64 Mahapatra A, Prasad T, Sharma T. Future J. Pharm. Sci. 2021; 7: 123
- 65 Mahdy H, Elnagar H, Sakr H. Al-Azhar J. Pharm. Sci. 2023; 67: 51
- 66 Li Z, Kou L, Fu X, Xie Z, Xu M, Guo L, Lin T, Gong S, Zhang S, Liu M. J. Enzyme Inhib. Med. Chem. 2022; 37: 1514
- 67 Akhtar W, Nainwal LM, Khan MF, Verma G, Chashoo G, Bakht A, Iqbal M, Akhtar M, Shaquiquzzaman M, Alam MM. J. Fluorine Chem. 2020; 236: 109579
- 68 Haoran W, Akhtar W, Nainwal LM, Kaushik SK, Akhter M, Shaquiquzzaman M, Alam MM. J. Heterocycl. Chem. 2020; 57: 3350
- 69 Xu Y, Hao SY, Zhang XJ, Li WB, Qiao XP, Wang ZX, Chen SW. Bioorg. Med. Chem. Lett. 2020; 30: 126885
- 70 Hu G, Wang C, Xin X, Li S, Li Z, Zhao Y, Gong P. New J. Chem. 2019; 43: 10190
- 71 Qin WW, Sang CY, Zhang LL, Wei W, Tian HZ, Liu HX, Chen SW, Hui L. Eur. J. Med. Chem. 2015; 95: 174
- 72 Kraljević TG, Klika M, Kralj M, Martin-Kleiner I, Jurmanović S, Milić A, Padovan J, Raić-Malić S. Bioorg. Med. Chem. Lett. 2012; 22: 308
- 73 Weinberg LR, Albom MS, Angeles TS, Husten J, Lisko JG, McHugh RJ, Milkiewicz KL, Murthy S, Ott GR, Theroff JP, Tripathy R. Bioorg. Med. Chem. Lett. 2011; 21: 164
- 74 Long L, Luo Y, Hou ZJ, Ma HJ, Long ZJ, Tu ZC, Huang LJ, Liu Q, Lu G. Eur. J. Med. Chem. 2018; 145: 805
- 75 Luo G, Tang Z, Lao K, Li X, You Q, Xiang H. Eur. J. Med. Chem. 2018; 150: 783
- 76 Zhao M, Ren H, Chang J, Zhang D, Yang Y, He Y, Qi C, Zhang H. Eur. J. Med. Chem. 2016; 119: 183
- 77 Jorda R, Havlíček L, Šturc A, Tušková D, Daumová L, Alam M, Škerlová J, Nekardová M, Peřina M, PospíšilT ŠJ. J. Med. Chem. 2019; 62: 4606
- 78 Jorda R, Havlícek L, McNae IW, Walkinshaw MD, Voller J, Šturc A, Navrátilová J, Kuzma M, Mistrík M, Bártek J, Strnad M. J. Med. Chem. 2011; 54: 2980
- 79 Wang R, Chen Y, Zhao X, Yu S, Yang B, Wu T, Guo J, Hao C, Zhao D, Cheng M. Eur. J. Med. Chem. 2019; 183: 111716
- 80 Sivaiah G, Raveesha R, Benaka Prasad SB, Yogesh Kumar K, Raghu MS, Alharti FA, Prashanth MK, Jeon B.-H. J. Mol. Struct. 2023; 1275: 134728
- 81 Alrooqi M, Khan S, Alhumaydhi FA, Asiri SA, Alshamrani M, Mashraqi MM, Alzamami A, Alshahrani AM, Aldahish AA. Anti-Cancer Agents Med. Chem. 2022; 22: 2775
- 82 Marinescu M, Popa CV. Int. J. Mol. Sci. 2022; 23: 5659
- 83 De S, Kumar S K A, Kumar Shah S, Kazi S, Sarkar N, Banerjee S, Deya S. RSC Adv. 2022; 12: 15385
- 84 Albratty M, Alhazmi HA. Arabian J. Chem. 2022; 15: 103846
- 85 Narasimhamurthy KH, Kallesha N, Mohan CD, Rangappa KS. Anticancer Functions of Pyridine Heterocycles . In Cytotoxicity - Understanding Cellular Damage and Response . Sukumaran A, Mansour MA. IntechOpen; London: 2023
- 86 El-Naggar M, Almahli H, Ibrahim HS, Eldehna WM, Abdel-Aziz HA. Molecules 2018; 23: 1459
- 87 Murthy IS, Sreenivasulu R, Alluraiah G, Ramesh Raju G. Russ .J. Gen. Chem. 2019; 89(8): 1718
- 88 Metwally NH, Deeb EA. Bioorg. Chem. 2018; 77: 203
- 89 Li X, Li S, Lu G, Wang D, Liu K, Qian X, Xue W, Meng F. Bioorg. Chem. 2020; 103: 104189
- 90 Wang R, Chen Y, Yang B, Yu S, Zhao X, Zhang C, Hao C, Zhao D, Cheng M. Bioorg. Chem. 2020; 94: 103474
- 91 Ning C, Tao A, Xu J. Eur. J. Med. Chem. 2023; 259: 115687
- 92 Gholap SS. Eur. J. Med. Chem. 2016; 110: 13
- 93 Akhtar J, Khan AA, Ali Z, Haider R, Shahar Yar M. Eur. J. Med. Chem. 2017; 125: 143
- 94 Petri GL, Spanò V, Spatola R, Holl R, Raimondi MV, Barraja P, Montalbano A. Eur. J. Med. Chem. 2020; 208: 112783
- 95 Kilic-Kurt Z, Bakar-Ates F, Aka Y, Kutuk O. Bioorg. Chem. 2019; 83: 511
- 96 Carta D, Bortolozzi R, Sturlese M, Salmaso V, Hamel E, Basso G, Calderan L, Quintieri L, Moro S, Viola G, Ferlin MG. Eur. J. Med. Chem. 2017; 127: 643
- 97 Kurup S, McAllister B, Liskova P, Mistry T, Fanizza A, Stanford D, Slawska J, Keller U, Hoellein A. J. Enzyme Inhibit. Med. Chem. 2018; 33: 74
- 98 El-Gamal MI, Oh CH. J. Enzyme Inhibit. Med. Chem. 2018; 33: 1160
- 99 Mesaros EF, Angeles TS, Albom MS, Wagner JC, Aimone LD, Wan W, Lu L, Huang Z, Olsen M, Kordwitz E, Haltiwanger RC, Landis AJ, Cheng M, Ruggeri BA, Ator MA, Dorsey BD, Ott GR. Bioorg. Med. Chem. Lett. 2015; 25: 1047
- 100 Narva S, Chitti S, Bala BR, Alvala M, Jain N, Kondapalli VG. C. S. Eur. J. Med. Chem. 2016; 114: 220
- 101 Williams IS, Joshi P, Gatchie L, Sharma M, Satti NK, Vishwakarma RA, Chaudhuri B, Bharate SB. Bioorg. Med. Chem. Lett. 2017; 27: 3683
- 102 Alanazi AS, Mirgany TO, Alsaif NA, Alsfouk AA, Alanazi MM. Saudi Pharm. J. 2023; 31: 989
- 103 Niedziałkowski P, Czaczyk E, Jarosz J, Wcisło A, Białobrzeska W, Wietrzyk J, Ossowski T. J. Mol. Struct. 2019; 1175: 488
- 104 Tikhomirov AS, Shtil AA, Shchekotikhin AE. Recent Pat. Anti-Cancer Drug Discovery 2018; 13: 159
- 105 Anifowose A, Agbowuro AA, Tripathi R, Lu W, Tan C, Yang X, Wang B. Med. Chem. Res. 2020; 29: 1199
- 106 Zheng Y, Zhu L, Fan L, Zhao W, Wang J, Hao X, Zhu Y, Hu X, Yuan Y, Shao J, Wang W. Eur. J. Med. Chem. 2017; 125: 902
- 107 Choudhary N, Collignon TE, Tewari D, Bishayee A. Phytomedicine 2022; 105: 154356
- 108 Dobson J, de Queiroz GF, Golding JP. Vet. J. 2018; 233: 8
- 109 Kwiatkowski S, Knap B, Przystupski D, Saczko J, Kędzierska E, Knap-Czop K, Kotlińska J, Michel O, Kotowski K, Kulbacka J. Biomed. Pharmacother. 2018; 106: 1098
- 110 Champeau M, Vignoud S, Mortier L, Mordon S. J. Photochem. Photobiol., B 2019; 197: 111544
- 111 Woźniak M, Nowak M, Lazebna A, Więcek K, Jabłońska I, Szpadel K, Grzeszczak A, Gubernator J, Ziółkowski P. Pharmaceuticals 2021; 14: 374
- 112 Banerjee SM, El-Sheikh S, Malhotra A, Mosse CA, Parker S, Williams NR, Macrobert AJ, Hamoudi R, Bown SG, Keshtgar MR. S. J. Clin. Med. 2020; 9: 483
- 113 Ostańska E, Aebisher D, Bartusik-Aebisher D. Biomed. Pharmacother. 2021; 137: 111302
- 114 Afanasiev MS, Dushkin AD, Grishacheva TG, Afanasiev SS, Karaulov AV. Photodiagn. Photodyn. Ther. 2022; 37: 102620
- 115 Warowicka A, Popenda Ł, Bartkowiak G, Musidlak O, Litowczenko-Cybulska J, Kuźma D, Nawrot R, Jurga S, Goździcka-Józefiak A. Phytomedicine 2019; 64: 152919
- 116 Nowak-Perlak M, Ziółkowski P, Woźniak M. Phytomedicine 2023; 119: 155035
- 117 Stanojković T, Marković V, Matić IZ, Mladenović MP, Petrović N, Krivokuća A, Petković M, Joksović MD. Bioorg. Med. Chem. Lett. 2018; 28: 2593
- 118 Wan Y, Fang G, Chen H, Deng X, Tang Z. Eur. J. Med. Chem. 2021; 226: 113837
- 119 Das R, Tambe G, Shard A. Results Chem. 2023; 5: 100950
- 120 Soliman AM, Alqahtani AS, Ghorab MM. J. Enzyme Inhibit. Med. Chem. 2019; 34: 1030
- 121 Luo Y, Li Y, Qiu K.-M, Lu X, Fu J, Zhu H.-L. Bioorg. Med. Chem. 2011; 19: 6069
- 122 Ghorab MM, Soliman AM, El-Adl K, Hanafy NS. Bioorg. Chem. 2023; 140: 106791
- 123 Cao L, Yao H, Yu L, Ren Y, Liu J, Li X, Jia X. Bioorg. Med. Chem. 2023; 85: 117241
- 124 Saleh NM, El-Gaby MS. A, El-Adl K, Abd El-Sattar NE. A. Bioorg. Chem. 2020; 104: 104350
- 125 Wang K, Song L.-H, Liang Q.-L, Zhang Y, Ma X.-L, Wang Q, Zhang H.-Y, Jiang C.-N, Wei J.-H, Huang R.-Z. Eur. J. Med. Chem. 2023; 254: 115349
- 126 Song J, Gao Q.-L, Wu B.-W, Li D, Shi L, Zhu T, Lou J.-F, Jin C.-Y, Zhang Y.-B, Zhang S.-Y, Liu H.-M. Eur. J. Med. Chem. 2019; 183: 111731
- 127 Reda Aouad M, Almehmadi MA, Faleh Albelwi F, Teleb M, Tageldin GN, Abu-Serie MM, Hagar M, Rezki N. Bioorg. Chem. 2022; 124: 105816
- 128 Swain B, Sahoo SK, Singh P, Angeli A, Yaddanapudi VM, Supuran CT, Arifuddin M. Eur. J. Med. Chem. 2022; 234: 114247
- 129 Liu Y, Wu Y, Sun L, Gu Y, Hu L. Eur. J. Med. Chem. 2020; 191: 112181
- 130 Luo Y, Zhou Y, Song Y, Chen G, Wang Y.-X, Tian Y, Fan W.-W, Yang Y.-S, Cheng T, Zhu H.-L. Bioorg. Med. Chem. Lett. 2018; 28: 3634
- 131 For a review see: Nayak S, Gaonkar SL, Musad EA, Dawsar AM. A. L. J. Saudi Chem. Soc. 2021; 25: 101284
- 132 Boström J, Hogner A, Llinàs A, Wellner E, Plowright AT. J. Med. Chem. 2012; 55: 1817
- 133 Naaz F, Ahmad F, Lone BA, Pokharel YR, Fuloria NK, Fuloria S, Ravichandran M, Pattabhiraman L, Shafi S, Shahar Yar M. Bioorg. Chem. 2020; 95: 103519
- 134 Tantak MP, Malik M, Klingler L, Olson Z, Kumar A, Sadana R, Kumar D. Bioorg. Med. Chem. Lett. 2021; 37: 127842
- 135 Bhatt P, Sen A, Jha A. ChemistrySelect 2020; 5: 3347
- 136 Vinayak A, Sudha M, Lalita KS. Dhaka Univ. J. Pharm. Sci. 2017; 16: 11
- 137 Khalil NA, Kamal AM, Emam SH. Biol. Pharm. Bull. 2015; 38: 763
- 138 Yousef TA, Alhamzani AG, Abou-Krisha MM, Kanthimathi G, Raghu MS, Kumar KY, Prashanth MK, Jeon BH. Heliyon 2023; 9: e20300
- 139 Cho S, Oh S, Um Y, Jung J.-H, Ham J, Shin W.-S, Lee S. Bioorg. Med. Chem. Lett. 2009; 19: 382
- 140 Kumar K, Pradines B, Madamet M, Amalvict R, Kumar V. Eur. J. Med. Chem. 2014; 86: 113
- 141 Karthik Kumar K, Prabu Seenivasan S, Kumar V, Mohan Das T. Carbohydr. Res. 2011; 346: 2084
- 142 de Carvalho da Silva F, de Souza MC. B. V, Frugulhetti II. P, Castro HC, Souza SL. de O, de Souza TM. L, Rodrigues DQ, Souza AM. T, Abreu PA, Passamani F, Rodrigues CR, Ferreira VF. Eur. J. Med. Chem. 2009; 44: 373
- 143 Costa MS, Boechat N, Rangel ÉA, de Carvalho da Silva F, de Souza AM. T, Rodrigues CR, Castro HC, Junior IN, Lourenço MC. S, Wardell SM. S. V, Ferreira VF. Bioorg. Med. Chem. 2006; 14: 8644
- 144 Lal K, Kaushik CP, Kumar A. Med. Chem. Res. 2015; 24: 3258
- 145 Bokor É, Docsa T, Gergely P, Somsák L. Bioorg. Med. Chem. 2010; 18: 1171
- 146 Fallah Z, Tajbakhsh M, Alikhani M, Larijani B, Faramarzi MA, Hamedifar H, Mohammadi-Khanaposhtani M, Mahdavi M. J. Mol. Struct. 2022; 1255: 132469
- 147 Al-Masoudi NA, Al-Soud YA. Tetrahedron Lett. 2002; 43: 4021
- 148 Shawish I, Barakat A, Aldalbahi A, Alshaer W, Daoud F, Alqudah DA, Al Zoubi M, Hatmal MM, Nafie MS, Haukka M, Sharma A, de la Torre BG, Albericio F, El-Faham A. Pharmaceutics 2022; 14: 145
- 149 Oggu S, Akshinthala P, Katari NK, Nagarapu LK, Malempati S, Gundla R, Jonnalagadda SB. Heliyon 2023; 9: e15935
- 150 Matore BW, Banjare P, Guria T, Roy PP, Singh J. Eur. J. Med. Chem. Rep. 2022; 5: 100058
- 151 Valente S, Trisciuoglio D, De Luca T, Nebbioso A, Labella D, Lenoci A, Bigogno C, Dondio G, Miceli M, Brosch G, Del Bufalo D, Altucci L, Mai A. J. Med. Chem. 2014; 57: 6259
- 152 Bondock S, Fadaly W, Metwally MA. Eur. J. Med. Chem. 2010; 45: 3692
- 153 Delmas F, Avellaneda A, Di Giorgio C, Robin M, De Clercq E, Timon-David P, Galy J.-P. Eur. J. Med. Chem. 2004; 39: 685
- 154 Hong S, Kim J, Yun S.-M, Lee H, Park Y, Hong S.-S, Hong S. J. Med. Chem. 2013; 56: 3531
- 155 Ke S, Wei Y, Yang Z, Wang K, Liang Y, Shi L. Bioorg. Med. Chem. Lett. 2013; 23: 5131
- 156 Kaplan-Ozen C, Tekiner-Gulbas B, Foto E, Yildiz I, Diril N, Aki E, Yalcin I. Med. Chem. Res. 2013; 22: 5798
- 157 Khan KM, Rahim F, Wadood A, Taha M, Khan M, Naureen S, Ambreen N, Hussain S, Perveen S, Choudhary MI. Bioorg. Med. Chem. Lett. 2014; 24: 1825
- 158 O’Brien SE, Browne HL, Bradshaw TD, Westwell AD, Stevens MF. G, Laughton CA. Org. Biomol. Chem. 2003; 1: 493
- 159 Oanh DT. K, Van Hai H, Park SH, Kim HJ, Han BW, Kim HS, Hong JT, Han SB, Nam NH. Bioorg. Med. Chem. Lett. 2011; 21: 7509
- 160 Rawat S, Rawat DS, Negi B. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2021; 60: 409
- 161 Rezki N, Almehmadi MA, Ihmaid S, Shehata AM, Omar AM, Ahmed HE. A, Aouad MR. Bioorg. Chem. 2020; 103: 104133
- 162 Subba Rao AV, Swapna K, Shaik SP, Lakshma Nayak V, Srinivasa Reddy T, Sunkari S, Shaik TB, Bagul C, Kamal A. Bioorg. Med. Chem. 2017; 25: 977
- 163 Diao PC, Lin WY, Jian XE, Li YH, You WW, Zhao PL. Eur. J. Med. Chem. 2019; 179: 196
- 164 Lee E, An Y, Kwon J, Kim KI, Jeon R. Bioorg. Med. Chem. 2017; 25: 3614
- 165 Xie X.-X, Li H, Wang J, Mao S, Xin M.-H, Lu S.-M, Mei Q.-B, Zhang S.-Q. Bioorg. Med. Chem. 2015; 23: 6477
- 166 Cao S, Cao R, Liu X, Luo X, Zhong W. Molecules 2016; 21: 6
- 167 Song J, Gao QL, Wu BW, Zhu T, Cui XX, Jin CJ, Wang SY, Wang SH, Fu DJ, Liu HM, Zhang SY, Zhang YB, Li YC. Eur. J. Med. Chem. 2020; 203: 112618
- 168 Osmaniye D, Levent S, Karaduman AB, Ilgın S, Zkay Y, Kaplancikli ZA. Molecules 2018; 23: 2033
- 169 Belal A, Abdelgawad MA. Res. Chem. Intermed. 2017; 43: 3859
- 170 Haider K, Shrivastava N, Pathak A, Prasad Dewangan R, Yahya S, Shahar Yar M. Results Chem. 2022; 4: 100258
- 171 Philoppes JN, Lamie PF. Bioorg. Chem. 2019; 89: 102978