

Subscribe to RSS
DOI: 10.1055/s-0042-1749375
Recurrence of Anaplastic Large Cell Lymphoma in the Frontal Lobe After Eleven Years of the Initial Diagnosis: Histopathological Findings and Prognosis
Recorrência de linfoma anaplásico de grandes células no lobo frontal onze anos após o diagnóstico inicial: Achados histopatológicos e prognósticoAuthors
Funding Statement The authors declare that they have received no funding regarding the performance of the present study.


Abstract
Anaplastic large cell lymphoma (ALCL) is a rare, high-grade, T-cell neoplasm classified into cutaneous primary, systemic primary ALK-positive (ALK+), systemic primary ALK-negative, or breast-implant associated. Secondary involvement of the central nervous system (CNS) by systemic primary ALK+ ALCL is a rare occurrence. We present a case of CNS involvement by ALK+ ALCL eleven years after diagnosis of the primary tumor in the thoracic vertebra. The anatomopathological examination confirmed the diagnosis of ALK+ ALCL. A brief review of the treatment and the clinical and pathological aspects is presented.
Resumo
O linfoma anaplásico de grandes células (LAGC) corresponde a uma neoplasia de alto grau rara, com imunofenótipo T, que podendo ser dividido em primário cutâneo, primário sistêmico ALK positivo (ALK+), primário sistêmico ALK negativo, e associado a próteses mamárias. Acometimento secundário do sistema nervoso central (SNC) por LAGC primário sistêmico ALK+ é uma rara entidade. Os autores apresentam um caso de acometimento do SNC por LAGC ALK+ onze anos após o diagnóstico do tumor primário em vértebra torácica. O exame anatomopatológico confirmou o diagnóstico de LAGC ALK+. Fez-se também uma breve revisão de aspectos clínicos e patológicos e tratamento.
Keywords
anaplastic large cell lymphoma - central nervous system - pathology - immunohistochemistry - prognosisPalavras-chave
linfoma anaplásico de grandes células - sistema nervoso central - patologia - imuno-histoquímica - prognósticoIntroduction
ALK-positive anaplastic large cell lymphoma (ALK+ ALCL) is a single entity defined by the World Health Organization (WHO) as a T-cell lymphoma composed of generally large, pleomorphic lymphoid cells with abundant cytoplasm, chromosomal translocation involving the ALK gene, and ALK and CD30 expression on surface membranes. Upon microscopic examination, the most common pattern is large cells with eccentric, horseshoe-shaped nuclei. However, there is great morphological variation, with reports of the following patterns: small cells, lymphohistiocytic, Hodgkin-like, multinucleated giant cells, and fusocellular. Anaplastic large cell lymphoma accounts for about 3% of non-Hodgkin lymphomas in adults, and for 10% to 20% of lymphomas in children.[1] The clinical presentation is generally characterized by advanced stages (III or IV) and associated with B symptoms. Lymph node and extranodal involvement are common, including sites such as the skin, bones, soft tissues, lungs, and liver. Involvement of the bone marrow occurs in about 30% of the cases analyzed by immunohistochemistry.[2] Primary involvement of visceral and central nervous systems (CNS) is rare, and CNS metastasis is even rarer.
We herein report a case of ALK+ ALCL involving the thoracic vertebra in a pediatric patient, with subsequent emergence of the same pathology in the right frontal lobe after eleven years of remission.
Case Report
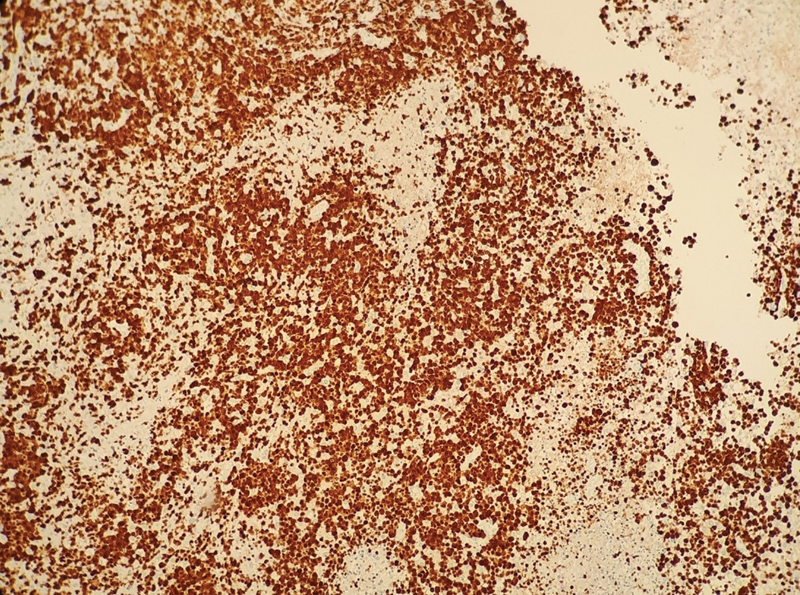
An 18-year-old patient presented to the hospital with nausea and vomiting followed by five generalized tonic-clonic seizures lasting 30 minutes, associated with salivation, tongue biting, and urinary incontinence, without recovery between the seizures. The patient was medicated with five ampoules of phenytoin and transferred to the reference hospital, with no further abnormalities on the neurological examination. A review of the medical records revealed a history of anaplastic lymphoma of the twelfth thoracic vertebra treated with chemotherapy. Eleven months of clinical follow-up showed no disease recurrence or involvement of the bone marrow, liver, spleen, tonsils, skin, or CNS. The patient underwent regular follow-up with the neurosurgery team for progressive vertebral collapse. Brain computed tomography (CT) scans of the last 8 years revealed the presence of an extensive area of encephalomalacia in the right frontal lobe, without significant changes between evaluations. During the current hospitalization, a magnetic resonance imaging (MRI) scan of the CNS ([Figs. 1] and [2]) was requested, which identified an expansive fronto-basal lesion on the right side, with marked low signal on fluid-attenuated inversion recovery (FLAIR) sequences and T1, hyperintense on FLAIR in the periphery, and a solid portion of blood-brain barrier breakdown near the lower anterior brain portion. The lesion measured about 4.8 × 3.3 cm in its largest diameters, and the solid portion with contrast enhancement measured about 1.3 cm in the largest diameter, which was suggestive of primary glial neoplasm. The patient underwent surgical intervention, with resection of the mural nodule, drainage of the cystic area via opening of the nodule/ventricle wall, and successful resolution of cerebrospinal fluid fistula, being discharged with outpatient follow-up. The anatomopathological examination revealed a poorly-differentiated malignant neoplasm compromising the CNS parenchyma, consisting of medium-sized round cells arranged in solid groups ([Figs. 3] and [4]) or in a perivascular pattern, with moderate mitotic index and areas of necrosis. The immunohistochemical examination showed that the neoplasm was positive for cluster determinant 2 (CD2), cluster determinant 3 (CD3), cluster determinant 5 (CD5), cluster determinant 30 (CD30) ([Fig. 5]), anaplastic lymphoma kinase (ALK) ([Fig. 6]), epithelial membrane antigen (EMA), perforin, and granzyme B, enabling the diagnosis of ALK+ ALCL involving the cerebral parenchyma. The proliferative index of the neoplasm was of about 90%, as estimated by Ki-67 expression ([Fig. 7]). The patient was referred to chemotherapy.






Discussion
Involvement of the brain parenchyma in cases of ALK+ ALCL is a rare event, with few reports of primary involvement[3] [4] [5] [6] [7] and even fewer of secondary involvement.[8] [9] [10] One study[11] analyzed the ALCL99 database, which comprises data on 618 patients with confirmed CNS biopsy between 1999 and 2017, involving 175 centers across 11 European countries and Japan. Central nervous system relapse was observed in 26 patients, 5 of whom had CNS disease at diagnosis. There was a mean interval of 8 months between the initial diagnosis and CNS relapse, with only 3 patients presenting relapse after 5 years; the latest relapse occurred 130.7 months after the initial diagnosis. We herein report the case of a patient who presented relapse in brain parenchyma 134 months after the initial diagnosis, becoming the latest case of relapse of ALK+ ALCL described in the literature.
The low incidence of CNS relapses can be explained by the effectiveness of the initial treatment and by the neoplasm's low tropism for the CNS. Central nervous system relapse is more common in patients who had the disease in this topography at the initial diagnosis, as well as in the case of bone marrow involvement or presence of peripheral blasts.[11] Patients with these risk factors could benefit from a more aggressive CNS prophylaxis.[11]
As reported, however, our patient did not have the indicated risk factors and had undergone a long asymptomatic period, raising the hypothesis of primary ALCL of the CNS or presence of viable neoplastic cells in the encephalomalacic area identified in CT scans in the initial years of follow-up, which were located at the same topographic site of the current neoplasm.
There is little agreement on the most appropriate therapeutic approach for CNS relapses. The most common treatment options include high-dose chemotherapy, autologous stem-cell transplantation, radiotherapy, and, recently, brentuximab vedotin (anti-CD30 drug).[12] [13] ALK-inhibiting drugs, such as crizotinib, ceritinib, and alectinib, were shown to promote long-lasting responses in patients with refractory and/or relapsed ALK+ ALCL. A clinical trial[14] used alectinib to treat ten patients, including children and older adults, with refractory/relapsed ALK+ ALCL; positive response was observed in eight out of the ten patients, and complete response, in six. Based on these results, in February 2020, Japan approved the use of this drug for refractory/relapsed ALK+ ALCL. Most clinical trials, however, exclude patients with CNS disease, explaining the lack of data on treatment efficacy in such cases. Vinblastine demonstrated good efficacy in relapsed ALCL, but there are few reports of the drug's efficacy when the tumor is located in the CNS, given the poor penetration of the blood-brain barrier. In fact, some patients had isolated CNS relapse despite their excellent systemic response.[15] [16]
New data on CNS penetration of second- and third-generation ALK inhibitors are emerging from studies on adults with ALK+ non-small cell lung carcinoma. Reports[17] [18] suggest that alectinib is superior to other ALK inhibitors with regard to CNS penetration (the penetration rates of crizotinib and alectinib were of 0.26% and 86% respectively[19]). Ceritinib, another ALK inhibitor, showed a rate of response of 75% in ALCL patients.[20] According to the International ALCL99 trial,[11] the 3-year survival rate of patients with CNS relapse is of 48.7%, suggesting that the goal of novel treatments should be to achieve complete cure.
In summary, we conclude that isolated CNS recurrence can arise even after more than a decade of the diagnosis and clinical remission of systemic ALK+ ALCL without primary CNS involvement. The need for prolonged follow-up becomes evident. Promising medications have emerged, and others are still in development; however, given the current heterogeneity in therapeutic approaches to the CNS relapse of ALCL, it is essential that new studies and strategies be developed.
Conflict of Interests
The authors have no conflict of interests to declare.
-
References
- 1 Stein H, Foss HD, Dürkop H. et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood 2000; 96 (12) 3681-3695
- 2 Fraga M, Brousset P, Schlaifer D. et al. Bone marrow involvement in anaplastic large cell lymphoma. Immunohistochemical detection of minimal disease and its prognostic significance. Am J Clin Pathol 1995; 103 (01) 82-89
- 3 Dong X, Li J, Huo N. et al. Primary central nervous system ALK-positive anaplastic large cell lymphoma in an adult: A rare case report. Medicine (Baltimore) 2016; 95 (49) e5534
- 4 Rupani A, Modi C, Desai S, Rege J. Primary anaplastic large cell lymphoma of central nervous system–a case report. J Postgrad Med 2005; 51 (04) 326-327
- 5 Vivekanandan S, Dickinson P, Bessell E, O'Connor S. An unusual case of primary anaplastic large cell central nervous system lymphoma: an 8-year success story. BMJ Case Rep 2011; 2011
- 6 Feng S, Chen Q, Chen J, Zheng P, Ma K, Tan B. Primary anaplastic large cell lymphoma of the central nervous system in a child: A case report. Medicine (Baltimore) 2020; 99 (29) e21115
- 7 Fujisawa E, Shibayama H, Mitobe F, Katada F, Sato S, Fukutake T. A case of primary central nervous system anaplastic lymphoma kinase positive anaplastic large cell lymphoma manifested as a unilateral pachymeningits. Rinsho Shinkeigaku 2017; 57 (11) 705-710
- 8 Mori T, Sugita K, Kimura K. et al. Isolated central nervous system (CNS) relapse in a case of childhood systemic anaplastic large cell lymphoma without initial CNS involvement. J Pediatr Hematol Oncol 2003; 25 (12) 975-977
- 9 Wang CX, Wang H, Li J. et al. Brain metastasis of ALK positive anaplastic large cell lymphoma after a long-term disease free survival in an old adult. Int J Clin Exp Pathol 2014; 7 (03) 1182-1187
- 10 Raffa EH, Branson HM, Ngan B, Alexander S, Abla O. Central nervous system relapse in a child with anaplastic large cell lymphoma: potential for new therapeutic strategies. Cancer Rep (Hoboken) 2021; 4 (05) e1377
- 11 Del Baldo G, Abbas R, Woessmann W. et al. Neuro-meningeal relapse in anaplastic large-cell lymphoma: incidence, risk factors and prognosis - a report from the European intergroup for childhood non-Hodgkin lymphoma. Br J Haematol 2021; 192 (06) 1039-1048
- 12 Pro B, Advani R, Brice P. et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 2012; 30 (18) 2190-2196
- 13 Pro B, Advani R, Brice P. et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2017; 130 (25) 2709-2717
- 14 Fukano R, Mori T, Sekimizu M. et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: An open-label phase II trial. Cancer Sci 2020; 111 (12) 4540-4547
- 15 Ruf S, Hebart H, Hjalgrim LL. et al. CNS progression during vinblastine or targeted therapies for high-risk relapsed ALK-positive anaplastic large cell lymphoma: A case series. Pediatr Blood Cancer 2018; 65 (06) e27003
- 16 Abid MB, Wang S, Loi HY, Poon LM. ALK-negative anaplastic large cell lymphoma with CNS involvement needs more than just brentuximab vedotin. Ann Hematol 2016; 95 (10) 1725-1726
- 17 Central nervous system relapse of systemic ALK-rearranged anaplastic large cell lymphoma treated with alectinib
- 18 Yang J, Li J, Gu WY. et al. Central nervous system relapse in a pediatric anaplastic large cell lymphoma patient with CLTC/ALK translocation treated with alectinib: A case report. World J Clin Cases 2020; 8 (09) 1685-1692
- 19 Wrona A. Management of CNS disease in ALK-positive non-small cell lung cancer: Is whole brain radiotherapy still needed?. Cancer Radiother 2019; 23 (05) 432-438
- 20 Schulte JH, Moreno L, Ziegler DS. et al. Final analysis of phase I study of ceritinib in pediatric patients with malignancies harboring activated anaplastic lymphoma kinase (ALK). Journal of Clinical Oncology 2020; 38: 10505
Address for correspondence
Publication History
Received: 10 June 2021
Accepted: 25 April 2022
Article published online:
05 September 2022
© 2022. Sociedade Brasileira de Neurocirurgia. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commecial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Thieme Revinter Publicações Ltda.
Rua do Matoso 170, Rio de Janeiro, RJ, CEP 20270-135, Brazil
-
References
- 1 Stein H, Foss HD, Dürkop H. et al. CD30(+) anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood 2000; 96 (12) 3681-3695
- 2 Fraga M, Brousset P, Schlaifer D. et al. Bone marrow involvement in anaplastic large cell lymphoma. Immunohistochemical detection of minimal disease and its prognostic significance. Am J Clin Pathol 1995; 103 (01) 82-89
- 3 Dong X, Li J, Huo N. et al. Primary central nervous system ALK-positive anaplastic large cell lymphoma in an adult: A rare case report. Medicine (Baltimore) 2016; 95 (49) e5534
- 4 Rupani A, Modi C, Desai S, Rege J. Primary anaplastic large cell lymphoma of central nervous system–a case report. J Postgrad Med 2005; 51 (04) 326-327
- 5 Vivekanandan S, Dickinson P, Bessell E, O'Connor S. An unusual case of primary anaplastic large cell central nervous system lymphoma: an 8-year success story. BMJ Case Rep 2011; 2011
- 6 Feng S, Chen Q, Chen J, Zheng P, Ma K, Tan B. Primary anaplastic large cell lymphoma of the central nervous system in a child: A case report. Medicine (Baltimore) 2020; 99 (29) e21115
- 7 Fujisawa E, Shibayama H, Mitobe F, Katada F, Sato S, Fukutake T. A case of primary central nervous system anaplastic lymphoma kinase positive anaplastic large cell lymphoma manifested as a unilateral pachymeningits. Rinsho Shinkeigaku 2017; 57 (11) 705-710
- 8 Mori T, Sugita K, Kimura K. et al. Isolated central nervous system (CNS) relapse in a case of childhood systemic anaplastic large cell lymphoma without initial CNS involvement. J Pediatr Hematol Oncol 2003; 25 (12) 975-977
- 9 Wang CX, Wang H, Li J. et al. Brain metastasis of ALK positive anaplastic large cell lymphoma after a long-term disease free survival in an old adult. Int J Clin Exp Pathol 2014; 7 (03) 1182-1187
- 10 Raffa EH, Branson HM, Ngan B, Alexander S, Abla O. Central nervous system relapse in a child with anaplastic large cell lymphoma: potential for new therapeutic strategies. Cancer Rep (Hoboken) 2021; 4 (05) e1377
- 11 Del Baldo G, Abbas R, Woessmann W. et al. Neuro-meningeal relapse in anaplastic large-cell lymphoma: incidence, risk factors and prognosis - a report from the European intergroup for childhood non-Hodgkin lymphoma. Br J Haematol 2021; 192 (06) 1039-1048
- 12 Pro B, Advani R, Brice P. et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 2012; 30 (18) 2190-2196
- 13 Pro B, Advani R, Brice P. et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2017; 130 (25) 2709-2717
- 14 Fukano R, Mori T, Sekimizu M. et al. Alectinib for relapsed or refractory anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: An open-label phase II trial. Cancer Sci 2020; 111 (12) 4540-4547
- 15 Ruf S, Hebart H, Hjalgrim LL. et al. CNS progression during vinblastine or targeted therapies for high-risk relapsed ALK-positive anaplastic large cell lymphoma: A case series. Pediatr Blood Cancer 2018; 65 (06) e27003
- 16 Abid MB, Wang S, Loi HY, Poon LM. ALK-negative anaplastic large cell lymphoma with CNS involvement needs more than just brentuximab vedotin. Ann Hematol 2016; 95 (10) 1725-1726
- 17 Central nervous system relapse of systemic ALK-rearranged anaplastic large cell lymphoma treated with alectinib
- 18 Yang J, Li J, Gu WY. et al. Central nervous system relapse in a pediatric anaplastic large cell lymphoma patient with CLTC/ALK translocation treated with alectinib: A case report. World J Clin Cases 2020; 8 (09) 1685-1692
- 19 Wrona A. Management of CNS disease in ALK-positive non-small cell lung cancer: Is whole brain radiotherapy still needed?. Cancer Radiother 2019; 23 (05) 432-438
- 20 Schulte JH, Moreno L, Ziegler DS. et al. Final analysis of phase I study of ceritinib in pediatric patients with malignancies harboring activated anaplastic lymphoma kinase (ALK). Journal of Clinical Oncology 2020; 38: 10505