
RSS-Feed abonnieren
DOI: 10.1055/s-0042-1755195
Novel and Practical Industrial Process Scale-Up of 5-Bromo-2-chloro-4-(methoxycarbonyl)benzoic acid, a Key Intermediate in the Manufacturing of Therapeutic SGLT2 Inhibitors


- Introduction
- Results and Discussion
- Conclusion
- Experimental Section
- Supporting Information
- References
Abstract
5-Bromo-2-chloro-4-(methoxycarbonyl)benzoic acid (1) is a key intermediate for the synthesis of a family of promising SGLT2 inhibitors currently in preclinical and phase I studies for diabetes therapy. In this investigation, cheap, easily available dimethyl terephthalate was used as the raw starting material, and compound 1 was prepared effectively in six steps, including nitration, hydrolysis, hydrogenation, esterification, bromination, and diazotization. The preparation was run successfully on approximately 70 kg/batch with the total yield of 24%. This practical process was demonstrated to be scalable with a great yield and significant cost reduction.
#
Keywords
5-bromo-2-chloro-4-(methoxycarbonyl)benzoic acid - SGLT2 inhibitors - practical process - scale upIntroduction
Diabetes is a serious, chronic disease that occurs either when the pancreas does not produce enough insulin (a hormone that regulates blood sugar, or glucose) or when the body cannot effectively use the insulin it produces. Diabetes is an important public health problem, one of four priority noncommunicable diseases targeted for action by world leaders. Both the number of cases and the prevalence of diabetes have been steadily increasing over the past few decades. Although there are antidiabetic drugs on the market, hyperglycemia is still clinically hard to be controlled. So, it is necessary to develop new antidiabetic drugs with novel targets and mechanisms. Sodium-glucose cotransporter-2 (SGLT2) inhibitors are a new class of diabetes treatment drugs. Mechanistically, they remove glucose from the urine by inhibiting the reabsorption of near-curved renal tubular glucose, which reduces blood glucose levels without depending on insulin. Besides, they could also reduce body weight, lower blood pressure, and have broad application prospects.[1]
In recent years, several research teams have devoted to the study of structure–activity relationship of SGLT2 inhibitors, and discovered that bromoaryls are active fragments to synthesize various promising candidate compounds as highly effective SGLT2 inhibitors.[2] [3] [4] [5] Among these inhibitors, 5-bromo-2-chloro-4-(methoxycarbonyl)benzoic acid (1) is a key intermediate in high demand that is utilized for developing SGLT-2 inhibitors ([Fig. 1]).[6] The known synthetic routes of compound 1 are all in small scale, in which the operation is complex and unpractical for industrial scale-up. Therefore, it is necessary to develop a feasible synthetic route and industrialized preparation method with high yield, low cost, and easy scale-up.

The reported synthetic route of compound 1 is shown in [Scheme 1].[7] [8] [9] Route A: 3-amino-4-toluic acid (2) is used as the starting material, the key intermediate 5 is obtained in three steps including aryl bromination, esterification, and Sandmeyer reaction. Route B: 2-bromo-4-methylbenzoic acid (6) goes through aromatic ring chlorination and esterification to prepare intermediate 5. The key intermediate 5 is oxidized with different oxidizing reagents and related conditions to obtain the target 1. These preparation methods have their deficits such as the expensive starting materials and undesirable oxidation efficiency, which is difficult to meet the market demand for industrialized mass production.

To develop an industrial process of intermediate 1, we conducted retro-synthesis analysis based on structural characteristics of the compound. Specifically, compound 1 can be synthesized from compound 12 through compound 11 ([Fig. 2]), based on the existing literature.[10] [11] [12] [13] Herein, a new synthetic route ([Scheme 2]) of compound 1 was designed and explored. Compound 11 is new compound that has not been reported in the literature.

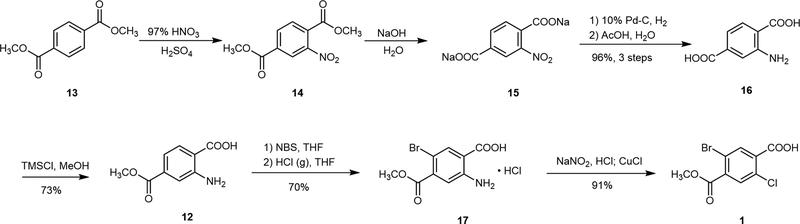
Utilizing cheap and commercially available dimethyl terephthalate (13) as a raw material, compound 14 was obtained by nitrification in the mixture of HNO3 and H2SO4. Then, 14 was hydrolyzed with NaOH and the product 16 is a solution of disodium salt. Direct catalytic hydrogenation of 15 without separation could easily provide 16, which reacted with TMSCl in MeOH to yield monoesterified intermediate 12. Compound 12 was brominated to obtain intermediate 11, which formed salt 17 (11·HCl) with hydrogen chloride gas. The target compound 1 was synthesized by the Sandmeyer reaction from compound 17.
After the optimization of the above synthetic route, 70 kg of compound 1 was prepared with high yield and purity, and this large-scale production is a feasible and low-cost new route.
#
Results and Discussion
In this new synthetic route of compound 1, compound 11 and its hydrochloride salt 17 are new compounds that have not been reported in the literature; it is the focus of this work. Compound 11 can be prepared by bromination of 12, but it also produces dibromo by-product 18 ([Scheme 3]), which is difficult to separate from compound 11 and unavoidably decreases the purity of final product. In this article, excess amount of N-bromosuccinimide (NBS) was used to react with compound 12 to prepare the main by-product 18, which was used as a reference substance for screening reaction conditions ([Scheme 4]). Both Br2 and NBS were used as brominating agents in tetrahydrofuran, respectively, and reaction conditions were investigated and optimized regarding the amounts of bromination reagents and reaction temperature ([Table 1]).


|
Entry |
Brominating agent |
Brominating agent (equiv.) |
Temp (°C) |
11 (%)[a] |
18 (%)[a] |
12 (%)[a] |
|---|---|---|---|---|---|---|
|
1 |
Br2 |
1 |
−20 to −10 |
92 |
1.1 |
5.5 |
|
2 |
Br2 |
1 |
−10 to 0 |
93 |
2.8 |
2.5 |
|
3 |
Br2 |
1 |
0–10 |
90 |
3.5 |
1.8 |
|
4 |
Br2 |
1.1 |
−20 to −10 |
95 |
3.2 |
1.1 |
|
5 |
NBS |
1 |
20–30 |
92 |
3.4 |
1.5 |
|
6 |
NBS |
1 |
10–20 |
91 |
3.0 |
2.6 |
|
7 |
NBS |
1 |
0–10 |
92 |
1.5 |
3 |
|
8 |
NBS |
1.1 |
0–10 |
93 |
1.7 |
1.5 |
|
9 |
NBS |
1.2 |
0–10 |
95 |
1.8 |
1.1 |
|
10 |
NBS |
1.22 |
0–10 |
96 |
1.8 |
0.8 |
a HPLC analysis. The conditions for HPLC method were: Thermo scientific-C18 column, C18 (5 μm, 150 mm × 4.6 mm); mobile phase A (0.1% H3PO4 in water) and B (CH3OH); 35:65 A/B→10:90 A/B, 25 minutes, 10:90 A/B, 10 minutes, 10:90 A/B→35:65 A/B, 1 minute, 35:65 A/B, 9 minutes; detection at 210 nm; flow rate = 1.0 mL/min.
The results showed that: (1) the proportion of dibromo impurity increased significantly as the reaction temperature increased ([Table 1], entries 1–7); (2) under the same equiv., NBS was a preferable brominating agent due to less formation of dibromo impurities 18 ([Table 1], entries 2 and 7); (3) using 1.22 equiv. of NBS gave the target compound 11 with the highest yield and purity ([Table 1], entries 7–10).
In summary, it was found that 1.22 equiv. of NBS was used as the brominating agent to carry out the reaction at 0–10°C and provided the best result. After the reaction, the solvent was removed by concentration under reduced pressure to obtain a mixed solid of product 11 and succinimide. The purification of 11 was achieved by refluxing the mixture in acetonitrile to remove succinimide because of its high solubility in acetonitrile.
Based on our knowledge, product 1 prepared from compound 11 by diazotization has not been reported in the literature. Theoretically, 1 could be prepared by diazotization, followed by Sandmeyer reaction. For the Sandmeyer reaction, the corresponding diazonium salt needed to be prepared first, and then the reaction was performed under the catalysis of cuprous salt to obtain the product ([Scheme 5]). Several commonly available industrial systems for the preparation of diazonium salts to generate compound 1 by Sandmeyer reaction were investigated.[14] [15] [16] The results showed that the yield of the reaction was relatively low (25–46%), which could not meet the requirement of the practical application of scale-up ([Table 2]).

|
Entry |
Acid system |
Yield (%) |
|---|---|---|
|
1 |
Conc. HCl |
30 |
|
2 |
Conc. H3PO4 |
25 |
|
3 |
Conc. H2SO4 |
40 |
|
4 |
Conc. HCl/AcOH |
35 |
|
5 |
Conc. H2SO4/AcOH |
46 |
a 1.15 equiv. of NaNO2 was used, and the reaction performed at −5 to 5°C, unless otherwise stated.
b CuCl/conc. HCl was used.
After carefully studying and analyzing the reaction process of diazonium–Sandmeyer, we found that the yield of the reaction was limited by the diazonium salt formation. The more of the diazonium salts was produced in the solvent, the higher yield would be obtained after work-up. Otherwise, the reaction would be mess if there were not sufficient diazonium salts formed in the process. As compound 11 was an electron-withdrawing group-substituted aniline, it was difficult to perform salinization thoroughly under the reaction conditions. Therefore, we need to design a route to fully salify 11 to improve the yield of the subsequent diazotization reaction. Considering that 11 was easily soluble in a certain volume of THF, we prepared 11 in THF and introduced hydrogen chloride gas to generate the hydrochloride 17. Intermediate 17 was poorly soluble in THF and could be collected by centrifugation from the solvent THF without further purification. Intermediate 17 was an ideal intermediate in the manufacturing process because it was chemical stable, free of moisture, and less hygroscopic.
After 17 was obtained, the conditions for the diazotization–Sandmeyer reaction with 17 were further screened and optimized ([Table 3]). It was found that when the 1.3 equiv. of NaNO2 was used, and the reaction temperature was −5 to 5°C, the final desired product 1 was obtained with the highest yield (92%).
|
Entry |
NaNO2 (equiv.) |
Yield (%) |
|---|---|---|
|
1 |
1.1 |
85 |
|
2 |
1.2 |
88 |
|
3 |
1.3 |
92 |
a The reaction was performed at −5 to 5°C, using conc. HCl as an acid system.
b CuCl/conc. HCl was used.
#
Conclusion
In this article, a novel process for the large-scale production of 1, a key intermediate of SGLT2 inhibitors, was developed. Using cheap and easily available dimethyl terephthalate (13) as the raw starting material, 1 was prepared in six steps, including nitration, hydrolysis, hydrogenation, esterification, bromination, and diazotization. The preparation scale was approximately 70 kg with the total yield of 24%. Compound 11 and its hydrochloride salt 17 are new compounds and chemically stable. Through process screening and optimization, the amount of dibromo impurity 18 produced in the bromination reaction of compound 11 was reduced significantly. The salt-forming reaction of intermediate 11 not only increased the stability of the corresponding compound, but also improved the yield of the subsequent diazotization reaction and Sandmeyer reaction (from 40 to 92%). This synthetic process is appealing in industry because it starts from cheap and easily available materials, avoids unfriendly oxidative procedure, and harvests high yield and purity of products.
#
Experimental Section
General
Unless otherwise specified, nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Biospin 400 MHz instrument using tetramethylsilane as the internal standard. All chemical shifts were reported in ppm. Mass spectrometry (MS) spectra were obtained on an Agilent 6460 QQQ mass spectrometer (Agilent, United States) analysis system. All materials were obtained from commercial suppliers and were used without further purification. Reactions' time and purity of the products were monitored by thin-layer chromatography (TLC) on FLUKA silica gel aluminum cards (0.2 mm thickness) with fluorescent indicator 254 nm. Column chromatography was run on silica gel (200–300 mesh) from Qingdao Ocean Chemicals (Qingdao, Shandong, China). Reaction progress and compound purity were determined by high-performance liquid chromatography (HPLC). The conditions of HPLC method were: Thermo scientific-C18 column, C18 (5 μm, 150 mm × 4.6 mm); mobile phase A (0.1% H3PO4 in water) and B (CH3OH), from 35:65 A/B to 10:90 A/B over 25 minutes, and keep 10:90 A/B over 10 minutes, from 10:90 A/B to 35:65 A/B over 1 minutes, and keep 35:65 A/B over 9 minutes; detection at 210 nm; flow rate = 1.0 mL/min.
#
Dimethyl 2-nitroterephthalate (14)
A dry and clean 1,000 L glass-lined reactor was charged with 98% H2SO4 (700 kg) and cooled to 10 to 15°C, and then 97% HNO3 (85.00 kg, 130.9 mol) was added carefully thereto at temperature not more than 30°C. After the addition, starting material 13 (200.00 kg, 103.0 mol) was added in portions. After the completion of the reaction confirmed by TLC (3:1, PE/EA) sampling, the mixture was added slowly into water (1,200 kg) at temperature not more than 30°C in another 2,000 L glass-lined reactor. The resulting mixture was stirred at 5 to 15°C for 0.5 hour, and the resulting slurry was filtered with a centrifuge and washed with water (600 kg) to give 14 (off-white solid), which was directly used in the next step without further purification.
#
Sodium 2-nitroterephthalate (15)
A clean 1,000 L glass-lined reactor was charged with water (580 kg) followed by the addition of NaOH (87.00 kg, 217.5 mol). The mixture was stirred until the solid dissolves completely. Then, the entire batch of 14 was added thereto and stirred at 80 to 85°C for 1 hour to give the solution of 15, which was directly used in the next step.
#
2-Aminoterephthalic Acid (16)
A clean and nitrogen-purged 1,000 L autoclave reactor was charged with 10% Pd/C (2.50 kg, wetted with ca. 60% water) under nitrogen, and then the solution of 15 was added thereto. After the addition, the reactor was evacuated and purged with N2 three times, then evacuated and purged with H2 three times. When the gas replacement was finished, the resulting mixture was purged with H2 to 0.5 to 1.0 MPa at 80 to 85°C until the end of the reaction. The reaction was cooled to 20 to 25°C, and the reactor was again evacuated and purged with N2 three times. The resulting reaction mixture was filtered with a titanium rod filter. The filtrate was acidized with AcOH/water (144 kg/400 kg) in another 1,500 L glass-lined reactor. The resulting slurry was filtered with a centrifuge. The filter cake was washed with water (100 kg), dried in a hot air circulation oven at 70 to 75°C until the water content is ≤0.5% to give 16 as a pale solid (179.1 kg, 96% yield from 13 to 16). ESI-MS (m/z): calcd. for C8H6NO4 − [M – H]− 180.0375, found: 180.04. 1H NMR (400 MHz, DMSO-d 6) δ 7.76 (d, J = 8 Hz, 1H, ArH), 7.30 (d, J = 4 Hz, 1H, ArH), 7.00 (dd, J = 8, 4 Hz, 1H, ArH); 13C NMR (100 MHz, DMSO-d 6) δ 172.85, 170.97, 168.75, 150.95, 136.90, 131.64, 117.47, 115.31.
#
2-Amino-4-(methoxycarbonyl)benzoic Acid (12)
A clean 1,500 L glass-lined reactor was charged with MeOH (480 kg), and then 16 (102.00 kg, 563.1 mol) and TMSCl (90.00 kg, 828.4 mol) were added thereto. After the addition, the reaction was heated to 57 to 63°C and held for 12 hours. When TLC (1:1, PE/EA) indicated the reaction was complete, approximately 320 kg solvent was distilled off under reduced pressure. Water (640 kg) and AcOEt (344 kg) were added thereto and stirred for 0.5 hour. Then, KHCO3 (153.00 kg, 1,528.3 mol) was added carefully and stirred for 0.5 hour. The resulting mixture was filtered with a PP filter. The filtrate was phase-separated in a 1,500 L glass-lined reactor, and the aqueous phase was acidized with AcOH/water (75 kg/128 kg) in another 1,500 L glass-lined reactor. The resulting slurry was filtered with a centrifuge. The filter cake was dried in a hot air circulation oven at 70 to 75°C until the water content is ≤0.5% to give 12 as a brownish-yellow solid (80.2 kg, 73%). ESI-MS (m/z): calcd. for C9H8NO4 − [M – H]− 194.0532, found: 194.06. 1H NMR (400 MHz, DMSO-d 6) δ 7.79 (d, J = 8 Hz, 1H, ArH), 7.41(d, J = 4 Hz, 1H, ArH), 7.03 (dd, J = 8, 4 Hz, 1H, ArH), 3.83 (s, 3H); 13C NMR (100 MHz, DMSO-d 6) δ 169.45, 166.48, 151.68, 134.32, 132.11, 117.84, 114.76, 113.42, 52.69.
#
2-Amino-5-bromo-4-(methoxycarbonyl)benzoic acid hydrochloride (17)
A dry and clean 1,000 L glass-lined reactor was charged with THF (617 kg), and then 12 (87.00 kg, 445.8 mol) was added. The mixture was cooled to 0 to 5°C. Then, NBS (97.59 kg, 548.3 mol) was added in portions thereto at temperature below 10°C. After the addition, the reaction was heated to 25 to 30°C and held for 3 hours. When HPLC indicated the reaction was complete, 470 kg solvent was distilled off under reduced pressure. The residue was solvent-exchanged and concentrated twice with MeCN (137 and 68 kg), and MeCN (87 kg) was added. The resulting mixture was stirred at 57 to 63°C for 0.5 hour, and then cooled to 5 to 10°C for 3 hours. The resulting slurry was filtered with a centrifuge. Another dry and clean 1,000 L glass-lined reactor was charged with THF (430 kg), and then the filter cake was added. Then the mixture was stirred until the solid dissolved. HCl (25 kg) was bubbled from the bottom of the reactor at temperature below 30°C and stirred for 1 hour. The resulting slurry was filtered with a centrifuge. The filter cake was washed with THF (50 kg), and air-dried to give 17 as a brown solid (96.9 kg, 70%). ESI-MS (m/z): calcd. for C9H7BrNO4 − [M – H]− 271.9637, found: 271.95, 273.95. 1H NMR (400 MHz, DMSO-d 6) δ 7.88 (s, 1H, ArH), 7.17 (s, 1H, ArH), 3.84 (s, 3H); 13C NMR (100 MHz, DMSO-d 6) δ 168.15, 166.44, 150.50, 136.98, 135.91, 119.14, 113.44, 102.20, 53.08.
#
5-Bromo-2-chloro-4-(methoxycarbonyl)benzoic Acid (1)
A clean 1,000 L glass-lined reactor was charged with 31% HCl (368 kg), and then 17 (80.00 kg, 257.6 mol) and water (140 kg) were added thereto. Then, the reaction was cooled to −5 to 0°C, and the solution of NaNO2 (23.6 kg, 342.0 mol) in water (140 kg) was added in portions thereto at temperature below 5°C. After the addition, the reaction was cooled to −5 to 5°C and held for 1 hour to give the diazonium salt solution. To another clean 2,000 L glass-lined reactor charged with 31% HCl (436 kg) was added CuCl (28.20 kg) thereto. Then, the diazonium salt solution was added slowly. The resulting mixture was stirred for 1 hour. The resulting slurry was filtered with a centrifuge. The filter cake was washed with water (150 kg), dried in a hot air circulation oven at 70 to 75°C until the water content is ≤0.5% to give 1 as an earth-yellow solid (68.8 kg, 91%). ESI-MS (m/z): calcd. for C9H5BrClO4 − [M – H]− 290.9138, found: 290.91, 292.91. 1H NMR (400 MHz, DMSO-d 6) δ 14.00 (s, 1H, COOH), 8.09 (s, 1H, ArH), 7.93 (s, 1H, ArH), 3.89 (s, 3H); 13C NMR (100 MHz, DMSO-d 6) δ 165.29, 165.00, 136.01, 135.74, 132.80, 131.28, 118.79 (C × 2), 53.54.
#
2-Amino-3,5-dibromo-4-(methoxycarbonyl)benzoic acid (18, Dibromide Impurity)
2-Amino-4-(methoxycarbonyl)benzoic acid (16) (1.95 g, 0.01 mol) was dissolved in THF and stirred for 10 minutes at 0°C, and then NBS (3.56 g, 0.02 mol) was added. The reaction mixture was stirred for 3 hours at room temperature. The reaction solution was concentrated. The residue was purified by column chromatography. ESI-MS (m/z): calcd. for C9H6Br2NO4 − [M – H]− 349.8724, found: 351.9. 1H NMR (400 MHz, DMSO-d 6) δ 11.06 (s, 1H, COOH), 7.97 (s, 1H, ArH), 7.09 (s, 2H, NH), 3.91 (s, 3H).
#
#
Supporting Information
Copies of NMR spectra and MS of compounds 12, 16, 17, 1, and 18 are included in the Supporting Information ([Figs. S1]—[14] [online only]).
#
#
Conflict of Interest
We declared no conflict of interest.
-
References
- 1 Manoj A, Das S, Kunnath Ramachandran A, Alex AT, Joseph A. SGLT2 inhibitors, an accomplished development in field of medicinal chemistry: an extensive review. Future Med Chem 2020; 12 (21) 1961-1990
- 2 Xu B, Lv B, Feng Y. et al. O-Spiro C-aryl glucosides as novel sodium-dependent glucose co-transporter 2 (SGLT2) inhibitors. Bioorg Med Chem Lett 2009; 19 (19) 5632-5635
- 3 Lv B, Xu B, Feng Y. et al. Exploration of O-spiroketal C-arylglucosides as novel and selective renal sodium-dependent glucose co-transporter 2 (SGLT2) inhibitors. Bioorg Med Chem Lett 2009; 19 (24) 6877-6881
- 4 Prous JR, Serradell N, Munoz R. et al. Preparation of pyrano[3,2-c]benzopyran-6(2h)-one derivatives as SGLT2 antidiabetic agents. WO Patent 2013045495 A1. April 2013
- 5 Liu H, Li J, Wang J. Process for preparation of C,O-spiro aryl glycoside compounds and use thereof. WO Patent 2016206604 A1. December, 2016
- 6 Kroh C, Lang IU, Rose H. et al. Use of sodium-dependent glucose transporter SGLT2 inhibitors for the prevention and/or treatment of cardiac diseases in felines. WO Patent 2021165177 A1. August, 2021
- 7 Chen YW, Feng Y, Xu BH. et al. Preparation of benzylic glycoside derivatives and their inhibitory effect on sodium-dependent glucose co-transporter SGLT. US Patent 20080242596 A1. October, 2008
- 8 Liu YH, Fu TM, Ou CY. et al. Improved preparation of (1S,3′R,4‘S,5′S,6’R)-5-chloro-6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-spiro[isobenzofuran-1(3H),2′-[2H]pyran]-3′,4′,5′-triol. Chin Chem Lett 2013; 24 (02) 131-133
- 9 Lee SH, Kim MJ, Lee SH, Kim J, Park HJ, Lee J. Thiazolylmethyl ortho-substituted phenyl glucoside library as novel C-aryl glucoside SGLT2 inhibitors. Eur J Med Chem 2011; 46 (07) 2662-2675
- 10 Qin ZW, Liu C, Shi YL. et al. Method for preparing 2-amino-4-methanesulfonamide methylbenzoate. CN Patent 103524386 A. January, 2014
- 11 Li Z, Zhang X, Zhang X. et al. 18F-Labeled benzyldiamine derivatives as novel flexible probes for positron emission tomography of cerebral β-amyloid plaques. J Med Chem 2016; 59 (23) 10577-10585
- 12 Skibo EB, Gilchrist JH. Synthesis and electrochemistry of pyrimidoquinazoline-5,10-diones. Design of hydrolytically stable high potential quinones and new reductive alkylation systems. J Org Chem 1988; 53 (18) 4209-4218
- 13 Ursuegui S, Yougnia R, Moutin S. et al. A biotin-conjugated pyridine-based isatoic anhydride, a selective room temperature RNA-acylating agent for the nucleic acid separation. Org Biomol Chem 2015; 13 (12) 3625-3632
- 14 Bauer E, Gerlach K, Pfau R. et al. Novel substituted nitrogen-containing heterobicycles and use thereof as factor Xa inhibitors. CA Patent 2511349 A1. July, 2004
- 15 Kim M, Boissonnault JA, Dau PV, Cohen SM. Metal-organic framework regioisomers based on bifunctional ligands. Angew Chem Int Ed Engl 2011; 50 (51) 12193-12196
- 16 Chen YW, Feng Y, Xu BH. et al. Glucose transport inhibitors and methods of use. US Patent 2007275907 A1. November, 2007
Address for correspondence
Publikationsverlauf
Eingereicht: 28. Februar 2022
Angenommen: 16. Juni 2022
Artikel online veröffentlicht:
22. August 2022
© 2022. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Manoj A, Das S, Kunnath Ramachandran A, Alex AT, Joseph A. SGLT2 inhibitors, an accomplished development in field of medicinal chemistry: an extensive review. Future Med Chem 2020; 12 (21) 1961-1990
- 2 Xu B, Lv B, Feng Y. et al. O-Spiro C-aryl glucosides as novel sodium-dependent glucose co-transporter 2 (SGLT2) inhibitors. Bioorg Med Chem Lett 2009; 19 (19) 5632-5635
- 3 Lv B, Xu B, Feng Y. et al. Exploration of O-spiroketal C-arylglucosides as novel and selective renal sodium-dependent glucose co-transporter 2 (SGLT2) inhibitors. Bioorg Med Chem Lett 2009; 19 (24) 6877-6881
- 4 Prous JR, Serradell N, Munoz R. et al. Preparation of pyrano[3,2-c]benzopyran-6(2h)-one derivatives as SGLT2 antidiabetic agents. WO Patent 2013045495 A1. April 2013
- 5 Liu H, Li J, Wang J. Process for preparation of C,O-spiro aryl glycoside compounds and use thereof. WO Patent 2016206604 A1. December, 2016
- 6 Kroh C, Lang IU, Rose H. et al. Use of sodium-dependent glucose transporter SGLT2 inhibitors for the prevention and/or treatment of cardiac diseases in felines. WO Patent 2021165177 A1. August, 2021
- 7 Chen YW, Feng Y, Xu BH. et al. Preparation of benzylic glycoside derivatives and their inhibitory effect on sodium-dependent glucose co-transporter SGLT. US Patent 20080242596 A1. October, 2008
- 8 Liu YH, Fu TM, Ou CY. et al. Improved preparation of (1S,3′R,4‘S,5′S,6’R)-5-chloro-6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-spiro[isobenzofuran-1(3H),2′-[2H]pyran]-3′,4′,5′-triol. Chin Chem Lett 2013; 24 (02) 131-133
- 9 Lee SH, Kim MJ, Lee SH, Kim J, Park HJ, Lee J. Thiazolylmethyl ortho-substituted phenyl glucoside library as novel C-aryl glucoside SGLT2 inhibitors. Eur J Med Chem 2011; 46 (07) 2662-2675
- 10 Qin ZW, Liu C, Shi YL. et al. Method for preparing 2-amino-4-methanesulfonamide methylbenzoate. CN Patent 103524386 A. January, 2014
- 11 Li Z, Zhang X, Zhang X. et al. 18F-Labeled benzyldiamine derivatives as novel flexible probes for positron emission tomography of cerebral β-amyloid plaques. J Med Chem 2016; 59 (23) 10577-10585
- 12 Skibo EB, Gilchrist JH. Synthesis and electrochemistry of pyrimidoquinazoline-5,10-diones. Design of hydrolytically stable high potential quinones and new reductive alkylation systems. J Org Chem 1988; 53 (18) 4209-4218
- 13 Ursuegui S, Yougnia R, Moutin S. et al. A biotin-conjugated pyridine-based isatoic anhydride, a selective room temperature RNA-acylating agent for the nucleic acid separation. Org Biomol Chem 2015; 13 (12) 3625-3632
- 14 Bauer E, Gerlach K, Pfau R. et al. Novel substituted nitrogen-containing heterobicycles and use thereof as factor Xa inhibitors. CA Patent 2511349 A1. July, 2004
- 15 Kim M, Boissonnault JA, Dau PV, Cohen SM. Metal-organic framework regioisomers based on bifunctional ligands. Angew Chem Int Ed Engl 2011; 50 (51) 12193-12196
- 16 Chen YW, Feng Y, Xu BH. et al. Glucose transport inhibitors and methods of use. US Patent 2007275907 A1. November, 2007