

Subscribe to RSS
DOI: 10.1055/s-0042-1757887
A Rare Biotinidase Deficiency in the Pediatrics Population: Genotype–Phenotype Analysis
Authors
Funding The Science and Engineering Research Board (SERB), Government of India (EMEQ/2019/000411) supported this work.

Abstract
Biotinidase (BTD) deficiency is a rare autosomal recessive metabolic disorder caused by insufficient biotin metabolism, where it cannot recycle the vitamin biotin. When this deficiency is not treated with supplements, it can lead to severe neurological conditions. Approximately 1 in 60,000 newborns are affected by BTD deficiency. The BTD deficiency causes late-onset biotin-responsive multiple carboxylase deficiency, which leads to acidosis or lactic acidosis, hypoglycemia, and abnormal catabolism. BTD deficiency is of two types based on the amount of BTD Enzyme present in the serum. A wide range of pathogenic mutations in the BTD gene are reported worldwide. Mutations in the BTD gene lead to profound and partial BTD deficiency. Profound BTD deficiency results in a severe pathogenic condition. A high frequency of newborns are affected with the partial deficiency worldwide. They are mostly asymptomatic, but symptoms may appear during stressful conditions such as fasting or viral infections. Several pathogenic mutations are significantly associated with neurological, ophthalmological, and skin problems along with several other clinical features. This review discusses the BTD gene mutation in multiple populations detected with phenotypic features. The molecular-based biomarker screening is necessary for the disease during pregnancy, as it could be helpful for the early identification of BTD deficiency, providing a better treatment strategy. Moreover, implementing newborn screening for the BTD deficiency helps patients prevent several diseases.
Keywords
biotinidase deficiency - BTD gene mutation - multiple carboxylase deficiency - neurological problemsIntroduction
Biotinidase (BTD) is an enzyme responsible for cleaving and recycling the biotin from biocytin and protein-bound sources.[1] [2] The biocytin is subsequently cleaved by BTD, which results in the release of biotin and lysine.[3] In humans, biotin, also known as vitamin B7 or Vitamin H, is water-soluble and is required for the coenzyme of five carboxylases, including amino acid metabolism, fatty acid metabolism, and gluconeogenesis. It is a proteolytic digestion product of holocarboxylase. BTD has biotinyl-transferase activity, where the biotin is transferred to histone from biocytin under physiological conditions.[4] BTD is a mammalian enzyme found primarily in serum, kidney, and liver.
BTD deficiency is a rare autosomal recessive metabolic disorder caused by insufficient biotin metabolism. In this condition, the body cannot appropriately recycle vitamin biotin. If it's not treated with supplements, it can lead to severe neurological conditions. BTD deficiency causes late-onset biotin-responsive multiple carboxylase deficiency (MCD), leading to acidosis or lactic acidosis, hypoglycemia, and abnormal catabolism.[5] [6] [7] The BTD deficiency was first reported by Wolf et al in 1981.[8] Approximately 1 in 60,000 newborns is affected with BTD deficiency.[9] The symptoms may occur after a few weeks of birth or in late childhood. In some conditions, the patient may be asymptomatic. The biotin supplement will reduce the symptoms. BTD deficiency is classified into two types: profound BTD deficiency and partial BTD deficiency, based on the BTD level present in the serum. The average amount of BTD in the human serum range is 4.4 to 10 nmol/min/mL, with a mean activity of 7.1 nmol/min/mL. Profound BTD deficiency means less than 10% of regular serum activity. A child with this type cannot recycle their endogenous biotin by cleaving it from biocytin, which causes MCD ([Fig. 1]). Untreated children have several neurological symptoms and clinical signs such as seizures, ataxia, hypotonia, hearing loss, vision problems, and developmental delay ([Fig. 2]). Unfortunately, they are irreversible.[2] [10] [11] Symptoms, including skin rashes, alopecia, conjunctivitis, ketolactic acidosis, and organic aciduria can be normalized when treated with supplements. Early treatment may help to prevent expression of symptoms. If untreated, the patient might die. Most symptoms of BTD deficiency appear between the ages of 1 week and 10 years with a mean age of 3.5 months.[8] Many countries are screening newborns at birth, to learn about prevalence of the BTD disorder and treat them with biotin supplementation. Partial BTD deficiency means 10 to 30% of BTD in the average serum activity in many populations. The patient with partial BTD deficiency is mostly asymptomatic ([Fig. 1]), symptoms may appear during stressful conditions such as fasting or viral infections.[12] A high level of BTD activity has been reported in children with glycogen storage disease type 1a, but the reason for the BTD level in this disease is unknown.[13]
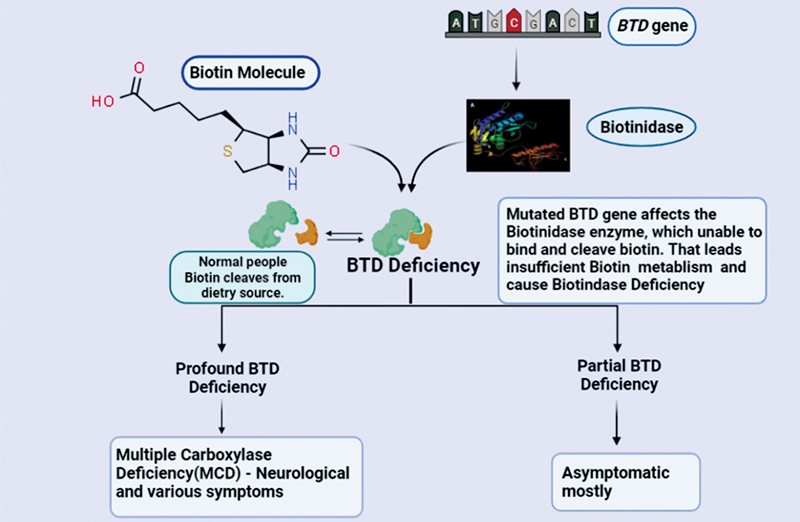

BTD deficiency fails to release biotin from dietary proteins, or endogenously synthesized carboxylase, which decreases bioavailability. Furthermore, urinary biotin excretion may increase due to renal filtration of free band impaired recycling of biotin from breakdown products of biotinylated carboxylases, such as biocytin.[14] It has a vital role in developing the central nervous system in humans. BTD is present in brain parts such as the red nucleus, lower auditory brainstem nuclei, and cerebellar Purkinje cells.[15] These are the reasons that can cause neurological problems among BTD deficiency patients. In addition, electrophysiological study helps identify the role of biotin deficiency in hearing loss. Studies show that a biotin-deficient diet results in prolonged latency on the auditory brainstem response test (ABR) or increased wave I-IV interpeak intervals as an evidence of a neurological finding.[16] The BTD deficiency patients' clinical symptoms varied, which are based on the severity of mutation.[17] [18]
Interestingly, United States and certain countries have mandatory newborn screening (NBS) programs that successfully help prevent or treat BTD deficiency symptoms by giving pharmacological doses of biotin supplements.[7] [17] The BTD deficiency is diagnosed among neonates. Enzymatic activity is measured by a colorimetric assay that uses biotinyl-p-aminobenzoate as a substrate.[19] Currently, BTD deficiency is primarily diagnosed by biochemical assay. The semiquantitative fluoroimmunoassay determines the BTD serum activity in the dried blood spots of neonates, which use biotinyl-6-aminoquinoline as a substrate.[20] Moreover, commercial ELISA kits are available to screen for BTD deficiency in newborns. If a positive result comes from the biochemical assay, we should further analyze the molecular test for confirmation. The mutations of the BTD gene have been detected by sequencing the gene. BTD gene mutation causes both profound and partial BTD deficiency in patients. The amino acid effect from BTD mutations determines the severity of the disease, either profound or partial BTD deficiency. Moreover, the D444H variant is the most common mutation in the BTD gene. This review discusses the BTD mutations in both profound and partial BTD deficiency among various populations with clinical signs, the prevalence of the BTD mutation, prevention of BTD deficiency through a NBS program, and significance of prenatal diagnosis.
BTD Gene
The BTD gene of humans (OMIM 609019) has been isolated and characterized.[21] [22] The BTD gene regulates the synthesis of BTD enzyme and is the only gene associated with BTD deficiency. It is located on chromosome 3q25.[21] The human BTD cDNA is from the cDNA hepatic library. The BTD cDNA encodes 543 amino acids, and their molecular weight is 61,133 Da. It has highly putative N-glycosylation sites and resultant total masses are approximately 74 and 80 kDa. In BTD, there are two potential AUG start codons and an open reading frame of 1629bp relative to the first AUG. The presence of an intron between the two possible start codons could allow alternative splicing. Both AUG start codons are in the same reading frame. Two start codons encode the two putative signal peptides, and the first peptide consists of 21 amino acids and the second of 41 amino acids. The BTD contains 13 cysteine residues, one of which is active and connects the biotinyl carboxyl group by a thiol ester before amide cleavage or biotinyl transfer.
Moreover, the human BTD gene contains four exons and three introns. The sizes of the exon 1, 2, 3, and 4 measure around 79 bp, 265 bp, 150 bp, and 1502 bp, respectively and the sizes of the respective intron regions are >12.5 kb, 6.2 kb, and 0.7 kb.[22] The first potential translation initiation codons are encoded in exon-1, while the second is encoded in exon-2. The nucleotide sequence upstream of exons 1 and 2 was examined for putative promoter elements. The promoter features identified from −600 to +400 are consistent with the ubiquitous expression of BTD, which has CpG island characteristics but lacks a TATA element. The six consensus methylation sites and three initiators (INR) sequences are considered necessary in the transcription initiation of TATA-less promoters. The consensus sequence for the HNF-5 liver-specific transcription factor is found at −352. The nucleotide sequence 5′ of exon 2, which contains the second putative ATG initiation codon, has features associated with housekeeping genes but does contain a consensus sequence for the liver-specific transcription factor C/EBP within 300 bp of the 5′ ends of exon 2.[11]
On a related note, when the human BTD amino acid sequence was compared with bacterial amidases and nitrolases, the result indicated that certain regions are highly conserved.[23] The conserved regions consist of active sites of cysteine of amidases and nitrolases that likely indicate the site of BTD involved in the thioester binding of biotin upon their cleavage from biocytin. In silico prediction tools and empiric data from the BTD enzyme activity help predict the pathogenicity of mutations, but sometimes the results are discordant. BTD gene sequencing is an essential tool for understanding the correlation between the genotype and biochemical phenotype of the patient.[23] [24]
BTD Mutation
Mutations in the BTD gene cause profound and partial BTD deficiency. More than 300 pathogenic mutations in the BTD gene have been identified, possibly causing BTD deficiency. Direct sequencing of some of these overlapping regions of the BTD gene, including intron and exon junctions, helps to perform mutation analysis. In BTD gene mutations, all types of mutations have been identified that can cause BTD deficiency. The mutations include missense, nonsense, cryptic splice site mutations, compound allelic mutations, single and multiple nucleotide insertions, single and multiple nucleotide deletions, and point mutations, which result from either premature stop codon formation or single amino acid substitution. Profound BTD deficiency arises in the homozygous or heterozygous state.[25] [26]
In addition, mutations have been identified in the entire coding sequence; however, none has been reported in exon 1. While several published studies have sequenced the entire BTD gene, including exon 1[25] [26] [27] [28] failure to find a mutation in exon 1 is most likely explained by the fact that exon 1 contains the first in-frame ATG, but not the highly conserved second ATG, which is the preferred or only actually used initiation codon. If so, we could not expect exon 1 changes to influence the BTD synthesis or secretion when the second ATG is the primary or only beginning site encoding the signal peptide sequence.[29] In some patients with BTD deficiency, there is no correlation between genotype and phenotype in BTD deficiency,[30] and BTD deficiency is primarily characterized based on skin and neurological abnormalities. The C-terminus of the protein mutation results in the severe loss of BTD activity.[25] Mutations in the carboxy-terminus of the BTD gene cause profound deficiency, and the site is affected by several missense-mutations.[23]
A high level of BTD gene mutations was identified in the U.S. population. Several unique mutations were reported in Turkey, France, United Kingdom, Saudi Arabia, Austria, Hungary, Italy, Brazil, and China. The BTD gene mutations are reported worldwide, including India, Germany, Pakistan, Sri Lanka, Afghanistan, Iraq, Poland, Nigeria, Iran, Spain, Sweden, Egypt, Syria, and Ethiopian populations, all listed in [Table 1]. According to the analysis of various studies and reports on BTD deficiency, a novel mutation in the BTD gene arises continuously every decade.
|
S. no |
cDNA |
Location |
Amino acid effect |
Phenotype |
Clinical symptoms |
Population |
Reference |
|---|---|---|---|---|---|---|---|
|
1 |
98–104del7ins3 |
Exon 2 |
Frameshift |
Patient with BTD |
N/A |
British |
[54] |
|
2 |
98_104del7ins3; 212T > C |
Exon 2; Exon 2 |
Frameshift |
Patient with BTD |
None |
Spain |
[25] |
|
3 |
99C > T |
Exon 2 |
C33C (PM) |
Patient with BTD |
None |
N/A |
[38] |
|
4 |
100G > A |
Exon 2 |
G34A |
Patient with BTD |
Neurological problems |
Spain |
[25] |
|
5 |
128A > G;1330G > C |
Exon 2; Exon 4 |
H43R |
Patient with BTD |
N/A |
United States |
[31] |
|
6 |
133G > A |
Exon 2 |
G45R |
Patient with BTD |
N/A |
Brazil |
[78] |
|
7 |
133G > A;865G > C |
Exon 2, Exon 4 |
G45R; A289P |
Patient with BTD |
N/A |
United States |
[30] |
|
8 |
133G > A;1271G > c |
Exon 2; Exon 4 |
G45R; C424S |
Patient with BTD |
Tachypnoea, eczema |
Scottish |
[38] |
|
9 |
133C > T |
Exon 2 |
H447Y |
Myelopathy (7-y-old boy) |
India |
[58] |
|
|
10 |
136G > T |
Exon 2 |
E46X |
Patient with BTD |
N/A |
Hungarian |
[56] |
|
11 |
159C > A; 160G > T |
Exon 2 |
H53Q; E54X |
Patient with BTD |
None |
Nigerian |
[38] |
|
12 |
171T > G |
Exon 2 |
Y57X |
Patient with BTD |
N/A |
Turkish |
[47] |
|
13 |
184G > T |
Exon 2 |
V62L |
Patient with BTD |
N/A |
United States |
[30] |
|
14 |
184G > A |
Exon 2 |
V62M |
Patient with BTD |
None |
Austria |
[26] |
|
15 |
190G > A |
Exon 2 |
E64K |
Patient with BTD |
N/A |
Spanish |
[28] |
|
16 |
192G > C;1330G > C |
Exon 2; Exon 4 |
E64D:D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
17 |
192–193insS |
Exon 4 |
L69H |
Patient with BTD |
N/A |
Turkish |
[43] |
|
18 |
194ins4 |
Exon 2 |
Frameshift |
Patient with BTD |
Hearing loss |
Turkish |
[37] |
|
19 |
194A > G |
Exon 2 |
H65R |
Patient with partial deficiency |
N/A |
Caucasian |
[52] |
|
20 |
203–206dup |
Exon 2 |
Frameshift |
Patient with BTD |
N/A |
Italy |
[62] |
|
21 |
211C > T |
Exon 2 |
L71L (PM) |
Patient with BTD |
N/A |
N/A |
[68] |
|
22 |
212T > C |
Exon 2 |
L71P |
Patient with BTD |
Asymptomatic |
N/A |
[38] |
|
23 |
212T > C;236G > A |
Exon 2 |
L71P; R79H |
Patient with BTD |
N/A |
Hungarian |
[57] |
|
24 |
235C > T |
Exon 2 |
R79C |
Patient with BTD |
N/A |
Turkish |
[47] |
|
25 |
235C > T;1361A > C |
Exon 2 |
R79C; Y454C |
Patient with BTD |
Sparse hair |
Turkish |
[38] |
|
26 |
235C > T;470G > A |
Exon 2; Exon 4 |
R79C; R157H |
Patient with BTD |
N/A |
United States |
[42] |
|
27 |
236G > A |
Exon 2 |
R79H |
Patient with BTD |
N/A |
Brazil |
[78] |
|
28 |
245C > A |
Exon 2 |
A82D |
Patient with BTD |
N/A |
Hungarian |
[57] |
|
29 |
245C > T;1330G > C |
Exon 2; Exon 4 |
A82V; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
30 |
245C > G |
Exon 2 |
A82G |
Patient with BTD |
N/A |
Turkish |
[79] |
|
31 |
246–254del9 |
Exon 2 |
L83-L85del |
Patient with BTD |
N/A |
N/A |
[54] |
|
32 |
248T > C |
Exon 2 |
L83S |
Patient with BTD |
N/A |
Egyptian |
[68] |
|
33 |
250G > C; 878dupT |
Exon3 |
Splice site; H294T |
Patient with profound deficiency |
Seizure, motor delay, dermatitis, and hearing loss |
Chinese |
[49] |
|
34 |
257T > G;1368A > c |
Exon 2; Exon 4 |
M86R; Q456H |
Patient with partial deficiency |
Mild sensorineural hearing loss. |
United States |
[5] |
|
35 |
262C > T |
Exon 2 |
Q88X |
Patient with BTD |
N/A |
N/A |
[54] |
|
36 |
278A > G |
Exon 2 |
Y93C |
Patient with BTD |
N/A |
Turkish |
[28] |
|
37 |
283C > T;1330G > C |
Exon 2; Exon 4 |
Q95X; D444H |
Patient with BTD |
None |
Caucasian |
[38] |
|
38 |
298G > A |
Exon 2 |
A100T |
Patient with BTD |
N/A |
Turkish |
[28] |
|
39 |
299C > T;1330G > C |
Exon 2; Exon 4 |
A100V; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
40 |
310G > T |
Exon 3 |
D104Y |
Patient with BTD |
N/A |
United States |
[30] |
|
41 |
321T > G;1330G > C |
Exon 3; Exon 4 |
I107M; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
42 |
326T > G |
Exon 3 |
V109G |
Patient with BTD |
N/A |
Austria |
[26] |
|
43 |
326dupT |
Exon 3 |
Frameshift |
Patient with BTD |
N/A |
United States |
[31] |
|
44 |
334G > C |
Exon 3 |
E112Q |
Patient with BTD |
N/A |
British |
[54] |
|
45 |
334G > A |
Exon 3 |
E112K |
Patient with BTD |
N/A |
Hungarian |
[57] |
|
46 |
341G > T |
Exon 3 |
G114V |
Patient with BTD |
UTI, hearing loss |
Syrian |
[38] |
|
47 |
356A > G;1459delT |
Exon 3 |
N119S |
Patient with BTD |
N/A |
United States |
[31] |
|
48 |
364A > G |
Exon 3 |
R122G |
Patient with BTD |
N/A |
United States |
[30] |
|
49 |
382T > G |
Exon 3 |
F128V |
Patient with BTD |
N/A |
Italy |
[33] |
|
50 |
383T > C |
Exon 3 |
F128S |
Patient with BTD |
N/A |
Turkish |
[79] |
|
51 |
386dupT |
Exon 3 |
Frameshift |
Patient with BTD |
N/A |
Italy |
[62] |
|
52 |
393delC |
Exon 3 |
Frameshift |
Patient with BTD |
N/A |
Palestinian |
[28] |
|
53 |
395T > G;637delC |
Exon 3; Exon 4 |
M132W; Frameshift |
Patient with BTD |
Hypotonia, fatigue, respiratory problems |
China |
[44] |
|
54 |
406delC |
Exon 3 |
Frameshift |
Neonates with BTD |
N/A |
Hungarian |
[55] |
|
55 |
419G > A |
Exon 3 |
W140 |
Patient with BTD |
N/A |
Turkish |
[43] |
|
56 |
420G > A;637delC |
Exon 3; Exon 4 |
W140X |
Patient with BTD |
Seizure, hypotonia |
China |
[44] |
|
57 |
424C > A |
Exon 3 |
P142T |
Patient with profound deficiency |
N/A |
Somalian |
[52] |
|
58 |
428G > T |
Exon 3 |
C143F |
Patient with BTD |
N/A |
Turkish |
[35] |
|
59 |
443G > A |
Exon 3 |
R148H |
Patient with BTD |
N/A |
N/A |
[68] |
|
60 |
444C > A |
Exon 3 |
A148A (PM) |
Patient with BTD |
N/A |
Spanish |
[80] |
|
61 |
445T > C |
Exon 3 |
F149L |
Patient with BTD |
N/A |
United States |
[30] |
|
62 |
449T > A |
Exon 4 |
V150G |
Patient with BTD |
Seizures, hypotonia |
Jordan |
[67] |
|
63 |
454A > C |
Exon 3 |
T152P |
Patient with BTD |
N/A |
Hungarian |
[56] |
|
64 |
455C > G;1330G > c |
Exon 3; Exon 4 |
T152R; D444H |
Patient with partial deficiency |
N/A |
United States |
[5] |
|
65 |
459G > A |
Exon 4 |
Splice site |
Patient with BTD |
N/A |
United States |
[54] |
|
66 |
460–1G > T |
Exon 4 |
V461D |
Patient with BTD |
Developmental delay |
China |
[81] |
|
67 |
464T > C;637delC |
Exon 4 |
L155P |
Patient with BTD |
Seizure, hypotonia |
China |
[44] |
|
68 |
466C > T |
Exon 4 |
Q156X |
Patient with BTD |
N/A |
Turkish |
[47] |
|
69 |
469C > T |
Exon 4 |
R157C |
Patient with BTD |
N/A |
Hungarian |
[56] |
|
70 |
470G > A |
Exon 4 |
R157H |
Patient with BTD |
N/A |
British |
[54] |
|
71 |
470G > A;1330G > C |
Exon 4 |
R157H; D444H |
Patient with BTD |
N/A |
United States |
[30] |
|
72 |
485C > T |
Exon 4 |
A162V |
Patient with BTD |
N/A |
United States |
[30] |
|
73 |
490–491del2 |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
Turkish |
[47] |
|
74 |
508G > A |
Exon4 |
V170M |
Patient with profound deficiency |
None |
Italy |
[63] |
|
75 |
511G > A:1330G > C |
Exon 4 |
A171T; D444H |
Patient with BTD |
N/A |
United States |
[30] |
|
76 |
515A > G |
Exon 4 |
N172S |
Patient with BTD |
None |
Austria |
[26] |
|
77 |
528G > T |
Exon 4 |
K176N |
Patient with BTD |
N/A |
United States |
[30] |
|
78 |
528–542del15 |
Exon 4 |
A197-S201 |
Patient with profound deficiency |
Seizures, hearing loss, hypotonia |
Iran |
[64] |
|
79 |
544delA |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
Turkish |
[47] |
|
80 |
557G > A |
Exon 4 |
C186Y |
Patient with BTD |
N/A |
Turkish |
[47] |
|
81 |
559C > T |
Exon 4 |
P187S |
Patient with BTD |
None |
Austria |
[25] |
|
82 |
566A > G; -34C > T |
Exon 4 |
D189G |
Patient with BTD |
N/A |
United States |
[31] |
|
83 |
582C > G;1330G > C |
Exon 4 |
F194L; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
84 |
583A > G |
Exon 4 |
N195D |
Patient with BTD |
N/A |
N/A |
[54] |
|
85 |
584A > G |
Exon 4 |
N195S |
Patient with BTD |
N/A |
Hungarian |
[56] |
|
86 |
587C > g |
Exon 4 |
196R |
Patient with BTD |
N/A |
Turkish |
[47] |
|
87 |
594–596del3 |
Exon 4 |
V199del |
Patient with BTD |
N/A |
N/A |
[54] |
|
88 |
594delC |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
Italy |
[62] |
|
89 |
595G > A |
Exon 4 |
V199M |
Patient with BTD |
N/A |
Brazil |
[78] |
|
90 |
605A > T |
Exon 4 |
N202I |
Patient with BTD |
N/A |
Ethiopian |
[68] |
|
91 |
617–619del/TTG |
Exon 4 |
V207del |
Patient with BTD |
N/A |
Turkish |
[79] |
|
92 |
625C > T;1368A > C |
Exon 4 |
R209C; Q456H |
Patient with BTD |
N/A |
United States |
[31] |
|
93 |
626G > A;1368A > C |
Exon 4 |
R209H; Q456H |
Patient with profound deficiency |
N/A |
United States |
5] |
|
94 |
629A > G |
Exon 4 |
Y210C |
Patient with BTD |
N/A |
British |
[54] |
|
95 |
631C > T |
Exon 4 |
R211C |
Patient with BTD |
N/A |
United States |
[30] |
|
96 |
631C > A |
Exon 4 |
R211S |
Patient with BTD |
NA |
Brazil |
[24] |
|
97 |
632G > A |
Exon 4 |
Splice site |
Neonates with BTD |
N/A |
Greek |
[18] |
|
98 |
632G > T |
Exon 4 |
R211L |
Patient with BTD |
N/A |
Brazil |
[24] |
|
99 |
641A > G;1330G > A |
Exon 4 |
N21AS; D444H |
Patient with BTD |
None |
Caucasian |
[38] |
|
100 |
643C > T |
Exon 4 |
L215F |
Patient with BTD |
N/A |
British |
[54] |
|
101 |
644T > A;637delC |
Exon 4 |
L215H |
Patient with BTD |
Fatigue, ataxia |
China |
[44] |
|
102 |
645C > T |
Exon 4 |
L215L (PM) |
Patient with BTD |
N/A |
N/A |
[68] |
|
103 |
646T > A;1330A > C |
Exon 4 |
Y216N; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
104 |
652G > C |
Exon 4 |
E218Q |
Patient with BTD |
N/A |
Hungarian |
[57] |
|
105 |
654G > C |
Exon 4 |
E218D |
Patient with BTD |
Respiratory problem, Hypotonia |
Caucasian |
[38] |
|
106 |
664G > C;1330G > C |
Exon 4 |
D222H; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
107 |
682G > T |
Exon 4 |
D228Y |
Patient with BTD |
N/A |
German |
[33] |
|
108 |
683A > G;1330G > C |
Exon 4 |
D228G; D444H |
Patient with partial deficiency |
N/A |
United States |
[5] |
|
109 |
692delC |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
United States |
[31] |
|
110 |
695T > C;1330G > C |
Exon 4 |
F232S; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
111 |
701C > T;1330A > G |
Exon 4 |
T234I; D444H |
Patient with partial deficiency |
N/A |
United States |
[5] |
|
112 |
709G > A;1330G > C |
Exon 4 |
A237T; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
113 |
731C > T |
Exon 4 |
T244I |
Patient with BTD |
N/A |
Turkish |
[35] |
|
114 |
734G > A |
Exon 4 |
C245Y |
Patient with BTD |
None |
Caucasian |
[38] |
|
115 |
743T > C;528G > T |
Exon 4 |
I248T; K176N |
Patient with BTD |
N/A |
United States |
[31] |
|
116 |
755A > G |
Exon 4 |
D262G |
Patient with BTD |
N/A |
United States |
[30] |
|
117 |
757C > T |
Exon 4 |
P253S |
Patient with BTD |
None |
Caucasian |
[38] |
|
118 |
758C > T;1489C > T |
Exon 4 |
P253L; P497S |
Patient with BTD |
N/A |
United States |
[31] |
|
119 |
764T > C |
Exon 4 |
I255T |
Patient with BTD |
N/A |
Swedish |
[68] |
|
120 |
770T > A;1330G > C |
Exon 4 |
V257D; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
121 |
783T > C;1330G > c |
Exon 4 |
Y261X; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
122 |
794A > T |
Exon 4 |
H265L |
Patient with BTD |
N/A |
Spanish |
[28] |
|
123 |
794A > T;933T > G |
Exon 4 |
H265L; S311R |
Patient with BTD |
N/A |
N/A |
[25] |
|
124 |
814T > G;1330G > C |
Exon 4 |
W272G; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
125 |
815G > A;1330G > C |
Exon 4 |
W272T; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
126 |
832C > G |
Exon 4 |
L278V |
Patient with BTD |
N/A |
Hungarian |
[57] |
|
127 |
833T > C |
Exon 4 |
L278P |
Patient with BTD |
N/A |
British |
[54] |
|
128 |
836T > G |
Exon 4 |
L279W |
Patient with BTD |
None |
Austria |
[26] |
|
129 |
836T > A;310–15delT |
Exon 4 |
L279X |
Patient with BTD |
N/A |
United States |
[31] |
|
130 |
856A > G;1330G > C |
Exon 4 |
K286E; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
131 |
858delA |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
Brazil |
[24] |
|
132 |
859G > A |
Exon 4 |
A287T |
Patient with BTD |
N/A |
Turkish |
[79] |
|
133 |
865G > C;1330G > C |
Exon 4 |
A289P; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
134 |
880A > G |
Exon 4 |
I294V |
Patient with BTD |
N/A |
United States |
[31] |
|
135 |
887T > G |
Exon 4 |
V296G |
Patient with BTD |
N/A |
German |
[28] |
|
136 |
895G > C;1413T > C |
Exon 4 |
A299P; C471C |
Patient with BTD |
N/A |
United States |
[31] |
|
137 |
896C > T |
Exon 4 |
A299V |
Patient with BTD |
N/A |
Spanish |
[80] |
|
138 |
898A > C;1330G > C |
Exon 4 |
N300H; D444H |
Patient with partial deficiency |
N/A |
United States |
[5] |
|
139 |
929G > A |
Exon 4 |
G310E |
Patient with BTD |
N/A |
Turkish |
[47] |
|
140 |
932G > A |
Exon 4 |
S311N |
Patient with BTD |
N/A |
United States |
[30] |
|
141 |
932G > C;1314T > A |
Exon 4 |
S311T; Y438X |
Patient with BTD |
N/A |
Brazil |
[24] |
|
142 |
933delT |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
French |
[54] |
|
143 |
933T > G; 933T > G |
Exon 4 |
S311R |
Patient with BTD |
N/A |
N/A |
[25] |
|
144 |
934G > A |
Exon 4 |
G313S |
Patient with BTD |
N/A |
Poland |
[28] |
|
145 |
935G > A |
Exon 4 |
G312D |
Patient with BTD |
N/A |
N/A |
[54] |
|
146 |
956C > T |
Exon 4 |
S319F |
Patient with BTD |
N/A |
Turkish |
[43] |
|
147 |
968A > G |
Exon 4 |
H323R |
Patient with BTD |
N/A |
Afghan |
[33] |
|
148 |
1001T > A;1330G > c |
Exon 4 |
I334N; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
149 |
1046A > C;1330G > C |
Exon 4 |
N349T; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
150 |
1049delC |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
Morocco |
[28] |
|
151 |
1052delC |
Exon 4 |
Frameshift |
Patient with BTD |
None |
Spain |
[25] |
|
152 |
1081T > G |
Exon 4 |
F361V |
Patient with BTD |
N/A |
Brazil |
[24] |
|
153 |
1096T > C |
Exon 4 |
S366P |
Patient with BTD |
N/A |
N/A |
[53] |
|
154 |
1096–1097dupTC |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
United States |
[31] |
|
155 |
1106C > T |
Exon 4 |
P368L |
Patient with BTD |
N/A |
Turkish |
[28] |
|
156 |
1126C > T;1612C > T |
Exon 4 |
Q376X; R538 |
Patient with BTD |
N/A |
United States |
[31] |
|
157 |
1157G > A |
Exon 4 |
W386X |
Patient with BTD |
N/A |
N/A |
[38] |
|
158 |
1158G > A |
Exon 4 |
W386X |
Patient with BTD |
N/A |
United States |
[30] |
|
159 |
1171C > T;1334G > T |
Exon 4; Exon 4 |
P391S (PM); G445V |
Patient with BTD |
Asymptomatic |
Caucasian |
[38] |
|
160 |
1191–1192del2 |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
N/A |
[28] |
|
161 |
1201G > A |
Exon 4 |
D401N |
Patient with BTD |
N/A |
Turkish |
[79] |
|
162 |
1205A > G |
Exon 4 |
N402S |
Patient with BTD |
N/A |
N/A |
[68] |
|
163 |
1207T > G;1330G > C |
Exon 4 |
F403V; D444H |
Patient with BTD |
N/A |
United States |
[30] |
|
164 |
1211C > T |
Exon 4 |
T404I |
Patient with BTD |
N/A |
Italy |
[62] |
|
165 |
1212–1222 del11 |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
Turkish |
[35] |
|
166 |
1214T > C |
Exon 4 |
L405P |
Patient with BTD |
N/A |
Swedish |
[68] |
|
167 |
1227–1241del15ins11 |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
United States |
[30] |
|
168 |
1239delC |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
N/A |
[28] |
|
169 |
1239del12 |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
N/A |
[28] |
|
170 |
1240–1251del12 |
Exon 4 |
414-V417del |
Patient with BTD |
N/A |
N/A |
[54] |
|
171 |
1241–1252del12bp |
Exon 4 |
Y414-V471del |
Patient with BTD |
N/A |
United States |
[31] |
|
172 |
1249G > T |
Exon 4 |
V417F |
Patient with partial deficiency |
N/A |
Caucasian |
[52] |
|
173 |
1250–1251TC > AG |
Exon 4 |
V417E |
Patient with BTD |
Asymptomatic |
China |
[81] |
|
174 |
1252T > C;1330G > C |
Exon 4 |
C418R; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
175 |
1253G > C |
Exon 4 |
C418S |
Patient with BTD |
N/A |
Hungarian |
[57] |
|
176 |
1264–1265insC |
Exon 4 |
Frameshift |
Patient with BTD |
Neurological problems |
Morocco |
[25] |
|
177 |
1267T > C |
Exon 4 |
C423R |
Patient with BTD |
N/A |
British |
[54] |
|
178 |
1268G > C |
Exon 4 |
C423W |
Patient with BTD |
None |
Austria |
[26] |
|
179 |
1271G > A |
Exon 4 |
C424Y |
Patient with BTD |
The respiratory problem, seizure |
Hispanic |
[38] |
|
180 |
1275T > G |
Exon 4 |
Y425X |
Patient with BTD |
None |
Austria |
[25] |
|
181 |
1284C > T;1489C > T |
Exon 4; Exon 4 |
Y4228Y(PM); P497S |
Patient with BTD |
Asymptomatic |
Nigerian |
[38] |
|
182 |
1284C > A |
Exon 4 |
Y428X |
Patient with BTD |
N/A |
China |
[27] |
|
183 |
1306G > A |
Exon 4 |
V471E |
Patient with BTD |
Asymptomatic |
China |
[81] |
|
184 |
1309C > G;1330G > C |
Exon 4 |
L437V; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
185 |
1313A > G |
Exon 4 |
Y438C |
Patient with BTD |
N/A |
Poland |
[28] |
|
186 |
1314T > A |
Exon 4 |
Y438X |
Patient with BTD |
N/A |
Brazil |
[78] |
|
187 |
1316T > C;1413T > C |
Exon 4 |
A439D; C471C |
Patient with profound deficiency |
Global developmental delay |
Sri Lankan |
[66] |
|
188 |
1320delG |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
Turkish |
[35] |
|
189 |
1328T > C |
Exon 4 |
F443S |
Patient with BTD |
N/A |
United States |
[31] |
|
190 |
1330G > C |
Exon 4 |
D444H |
Patient with BTD |
N/A |
Hungarian |
[33] |
|
191 |
1333G > A |
Exon 4 |
G445R |
Patient with BTD |
N/A |
Iraq |
[68] |
|
192 |
1334G > T |
Exon 4 |
G445V |
Patient with BTD |
N/A |
British |
[54] |
|
193 |
1134G > A;1330G > C |
Exon 4 |
G445E; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
194 |
1339C > T |
Exon 4 |
H447Y |
Spinal cord disease |
Seizures, Hypotonia |
United States |
[40] |
|
195 |
1352G > A |
Exon 4 |
G451D |
Patient with BTD |
N/A |
Afghan |
[33] |
|
196 |
1352–1353delGC |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
United States |
[31] |
|
197 |
1368A > C |
Exon 4 |
Q456H |
Patient with BTD |
N/A |
United States |
[30] |
|
198 |
1369G > A |
Exon 4 |
V457M |
Patient with BTD |
N/A |
Latin |
[54] |
|
199 |
1369G > C |
Exon 4 |
V457C |
Patient with BTD |
N/A |
Turkish |
[35] |
|
200 |
1372–1373insT |
Exon 4 |
C458fs |
Patient with partial defeciency |
United States |
[5] |
|
|
201 |
1382T > A;460–1G > T |
Exon 4 |
V461D |
Patient with BTD |
Developmental delay |
China |
[81] |
|
202 |
1384delA |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
China |
[27] |
|
203 |
1388G > A |
Exon 4 |
C463Y |
Patient with BTD |
N/A |
German |
[28] |
|
204 |
1413T > C |
Exon 4 |
C471C(PM) |
Patient with BTD |
N/A |
N/A |
[53] |
|
205 |
1432G > C |
Exon 4 |
A478P |
Patient with profound deficiency |
N/A |
Pakistan, India |
[52] |
|
206 |
1432G > A;755A > G |
Exon 4 |
A478T; D252G |
Patient with BTD |
N/A |
United States |
[31] |
|
207 |
1438G > A |
Exon 4 |
G480R |
Patient with BTD |
N/A |
Turkish |
[35] |
|
208 |
1455C > G;1330G > C |
Exon 4 |
H485Q; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
209 |
1457T > A |
Exon 4 |
L486Q |
Hyperkeratosis |
Eczematous lesions, hyperkeratotic erythema |
China |
[48] |
|
210 |
1458delG;278A > G |
Exon 4 |
Frameshift; Y93C |
Patient with BTD |
N/A |
United States |
[31] |
|
211 |
1459T > C |
Exon 4 |
W487R |
Patient with BTD |
N/A |
Turkish |
[47] |
|
212 |
1459delT |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
United States |
[31] |
|
213 |
1463G > A |
Exon 4 |
G488D |
Patient with BTD |
N/A |
Poland |
[54] |
|
214 |
1466A > C |
Exon 4 |
N489T |
Patient with BTD |
Asymptomatic |
Japan |
[53] |
|
215 |
1471A > G |
Exon 4 |
S491H |
Patient with BTD |
N/A |
Turkish |
[79] |
|
216 |
1475C > T |
Exon 4 |
T492I |
Patient with BTD |
Mild hypotonia |
Italy |
[60] |
|
217 |
1481A > G |
Exon 4 |
Y494C |
Patient with BTD |
N/A |
United States |
[31] |
|
218 |
1489C > T |
Exon 4 |
P497S |
Patient with BTD |
N/A |
United States |
[30] |
|
219 |
1493dupT;235C > T |
Exon 4; Exon 2 |
Frameshift; R79C |
Patient with BTD |
Seizures, fatigue, rash |
China |
[44] |
|
220 |
1493–1494insT |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
China |
[27] |
|
221 |
1511T > A |
Exon 4 |
M504K |
Patient with BTD |
N/A |
Hungarian |
[57] |
|
222 |
1526C > G;1330G > C |
Exon 4 |
P509R; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
223 |
1531G > A |
Exon 4 |
Q511E |
Patient with BTD |
None |
Caucasian |
[38] |
|
224 |
1595C > T |
Exon 4 |
T532M |
Patient with BTD |
N/A |
United States |
[30] |
|
225 |
1601C > T |
Exon 4 |
A534V |
Patient with BTD |
N/A |
Brazil |
[24] |
|
226 |
1610C > A |
Exon 4 |
G537V |
Patient with BTD |
N/A |
French |
[28] |
|
227 |
1612C > T |
Exon 4 |
R538C |
Patient with BTD |
N/A |
United States |
[54] |
|
228 |
1612C > A |
Exon 4 |
R538S |
Patient with BTD |
N/A |
United States |
[31] |
|
229 |
1613G > A;1062G > A |
Exon 4 |
R538H; T354T |
Patient with BTD |
N/A |
United States |
[31] |
|
230 |
1616–1617insT |
Exon 4 |
Frameshift |
Patient with BTD |
N/A |
United States |
[30] |
|
231 |
1619A > G |
Exon 4 |
Y540C |
Patient with BTD |
N/A |
United States |
[30] |
|
232 |
1627G > C |
Exon 4 |
D543H |
Patient with BTD |
None |
Turkish |
[26] |
|
233 |
1628A > T |
Exon 4 |
D543V |
Patient with BTD |
N/A |
United States |
[31] |
|
234 |
1629C > A;1330G > C |
Exon 4 |
D543E; D444H |
Patient with BTD |
N/A |
United States |
[31] |
|
235 |
44 + 1G > A |
Intron 1 |
Splice variant |
Patient with partial deficiency |
Asymptomatic |
United States |
[82] |
|
236 |
310–15delT |
Intron 2 |
mRNA expression |
Patient with partial deficiency |
N/A |
United States |
[5] |
|
237 |
12236G > A |
Intron |
Intronic |
Patient with BTD |
None |
Turkish |
[26] |
|
238 |
Entire Exon 1 Deletion |
Exon 1 |
N/A |
Patient with profound deficiency |
Seizure, Global developmental delay |
Sri Lanka |
[66] |
|
239 |
D444Y |
Patient with BTD |
N/A |
Italy |
[62] |
||
|
240 |
W487X |
Patient with BTD |
N/A |
Italy |
[62] |
||
Abbreviation: N/A, not available.
Mutation in the U.S. Population
In 2016, Procter et al discovered 48 novel mutations in a patient with BTD deficiency in the U.S. population. Their study analyzed 300 samples to determine the genotype to confirm the degree of BTD deficiency. The heterozygous novel alteration was found in all cases, and almost all of them have a second heterozygous mutation; such alterations are determined as mutations and classified as pathogenic. A total of 32 of 48 individuals had a second mutation with D444H in another allele. Children with partial BTD deficiency mostly had a D444H second mutation in another allele. One of the individuals had a missense alteration in one allele and an alteration within an intron in another allele. The alteration in the intron does not affect the enzymatic activity.[31]
A transversion of the 1368A > C cDNA mutation causes the substitution of histidine for glutamine at 456 positions. The Q456H amino acid substitution is a simple polymorphism and is the most common mutation in BTD deficiency in children identified in NBS.[32] The partial BTD deficiency usually occurs when an individual has one allele that results in nearly total loss of activity in combination with an allele having the D444H mutation. The D444H variant of BTD deficiency is similar to the Duarte variant in galactosemia.[33]
Cowan et al studied the BTD deficiency in California from July 2007 to June 2011. The population had profound BTD deficiency being estimated at one in 73,629 and profound plus partial variant cases at one in 31,717. Compared with global incidence, California has demonstrated a higher ratio of patients with one in 112,271 and one in 60,089 with profound variant and combined cases, respectively.[34]
Fascinatingly, Li et al identified the first intronic mutation in the BTD gene c.310–15delT in the U.S. population. The homozygous mutation p.T234I found in a child, which causes profound deficiency, also carries a single benign polymorphism, p.C471C. The combination of p.M86R with p.Q456H mutation is pathogenic. The p.Q456H is the other most common mutation that is combined with another mutation in the second allele.[5]
However, partial BTD deficiency in patients with spinal cord disease had a novel missense mutation, H447Y, which also caused the profound BTD deficiency. The individual also had mild alopecia, rapidly progressive symptoms, atypical neurological features, and an absence of cutaneous manifestations that initially led to an erroneous diagnosis and treatment. In the United States , the four mutations most commonly linked with complete BTD deficiency are C33Ffs*36, Q456H, R538C, and the double mutation D444H: A171T. Partial BTD deficiency is almost universally accredited to the D444H mutation.[11]
Mutation in the Turkish Population
BTD deficiency is higher in Turkey compared with the rest of the world. The recently published data from the Ministry of Health, Turkey, reported the incidence at approximately one in 7,116, compared with worldwide incidence of one in 60,000. Karaca et al study of mutations in NBS found 26 mutations in 192 patients, where 91% of the BTD patients were affected with two mutations in two different alleles. The p.R157H, p.D444H, c.98–104del7ins3, and p.T532M were commonly found in 72.3% of the mutant alleles.[35] Mutations R79C, Y93C, and R211F create a new abnormal cysteine, while C143F, C186Y, and C418S mutations reveal a loss of cysteine residues in the BTD gene. There are 13 functionally important cysteine residues present in the matured BTD enzyme.[21]
Various studies suggest that the mutations T196R, V199M, G310E, P368L, Y454C, R538C, D543H, and 3′ splice site mutation with 100G > A are the most common mutations in the Turkish population.[6] [26] [36] [37] [38] D444H, H447Y, Q456H, V457L, V457M, G480R, P497S, and T532M mutations are located in a conserved region that corresponds to the protein's C-terminus and may impact its biotin-binding properties.[29] [36] [39]
In Turkey, NBS program is mandatory to detect BTD deficiency. Interestingly, mutation H447Y is associated with myelopathy, whose amino acid effect disrupts the nearby disulfide bond formation.[40] In the southeastern part of Turkey, there is a high rate of consanguineous marriages. The children of these parents were affected with homozygous or compound heterozygous mutations (c.235C > T in exon 2 and c.470G > A, c.557G > A, c.1330G > C, c1368A > C, c1489C > T, and c1595C > T mutations in exon 4), which are known to cause profound BTD deficiency.[41] These results were gathered from the study of BTD-deficient families. BTD mutation affected around 85% of the families screened. The father, mother, and siblings with BTD deficiency are studied in this family study. The family studies of the BTD patients are also important to detect this disorder. Most family members might be asymptomatic and transfer this disorder to their children.[42]
In one of the studies, a patient under biotin supplementation in Turkey had a clinical presentation of metabolic acidosis and ketosis during an acute gastroenteritis episode. The above patient had a homozygous mutation of W140X.[43] and a 16-year-old girl was diagnosed with partial BTD insufficiency. She suffered from recurring hypoglycemia and ketoacidosis crises, most frequently during upper respiratory tract illnesses. She had c98–104delinsTCC and D444H compound heterozygous mutations. Biotin therapy helped her to become asymptomatic. One of the patients with profound deficiency had sudden vision loss and muscular weakness, and he was treated with steroids before the diagnosis.[43] The symptoms started getting better post the oral supplementation of biotin.
Mutation in the Chinese Population
The BTD gene mutations are rarely reported among the Chinese population. In southern China, the BTD mutation is analyzed in BTD patients by Liu et al. They detected 10 different mutations; five were previously reported, such as R79C, C424S, C471Y, R538H, and H213TfsTer51. They found another five novel mutations such as M132W, L155P, L215H, W140X, and L498FfsTer13. Approximately, 68.75% of these mutations were localized in exon 4, which contains the enzyme active sites and is a hot spot for mutations.[36] Chinese patients with BTD deficiency were misdiagnosed as having encephalopathy, myelitis, or dermatitis at the onset.[44] Spinal cord impairment is a normal manifestation of delayed-onset BTD deficiency and is uncommon to recognize.[45] [46] Therefore, earlier detection is vital to prevent those severe illnesses and saving a patient's life.
In the Chinese population G98: d7i3, R538C, and Q456H are the most common mutations that appear to be the hot spot mutations for profound BTD variants reported in other countries.[47] Ye et al reported five different mutations in four patients with BTD deficiency from northern China,[27] of which only c.1493dupT was also detected in Liu et al study. The results indicate that the mutation spectrum of BTD deficiency may differ in different countries, even different districts in China. In China, the patient with profound BTD deficiency had several common clinical symptoms such as hypotonia, fatigue, hearing deficits, skin rash, proximal muscle weakness, respiratory problems, and seizures. In vitro study of the D444H mutation reduces the protein expression and does not affect BTD enzyme activity.[44] The incidence of BTD is unknown in China; only large city hospitals have NBS programs for BTD. A girl in China had clinical symptoms in skin and hair associated with BTD deficiency. She has been affected by hyperkeratosis in her hands, feet, and blonde hair, and her grandparent was consanguineous. The proband's biotin level was 0.048 pmol/min mm3 by dry filter paper blood smear.[48] The consanguineous marriage might influence this inherited disorder.
Recently, a 17-year-old female with a profound deficiency in China exhibited novel BTD gene heterozygous variants, c.250-1G > C and c.878dupT, were identified. The mutation c.250-1G > C was inherited from her father, and c.878dupT was inherited from her mother. The patient showed various phenotypic characteristics, such as eczema-like rash, hair loss, hearing loss, hypotonia, and spontaneous recurrent epilepsy.[49]
Mutations in Other Populations
In BTD patients, the alopecia is a common manifestation among Iranian and Indian populations (eight in 16 and nine in 10, respectively).[50] [51] Minnesota is a U.S. state, where 40,000 Somalia immigrant people and their children live. Minnesota has a high incidence of combined profound and partial BTD deficiency of one in 8,540, and the profound incidence is one in 52,945, which is unusually high compared with a report worldwide. Homozygous mutation P497S was found in Somalia patients whose parents are consanguineous married[52]; that same mutation is also reported in the Caucasian population.[29] The mutation A478P was found in individuals of Asian ethnic background, affected with profound BTD deficiency.
Four recurrent mutations most frequently cause profound deficiency. In symptomatic patients, mutation G98: d7i3 and R538 are found with high frequency, while in asymptomatic patients, Q456H missense mutation and A171T were frequent. D444H mutation with combined variant was found with high frequency during NBS.[32] [53] [54] A study in western Hungary found that Q456H (14.2%), A171T (?), D444H (10%), c.98_104del7insTCC (7.5%), and R538C (5%) mutations were common among children with BTD deficiency.[55] A newborn screening in western Hungary population found BTD deficiency in 57 patients among 1,070,000 neonates from the year 1989 to 2008. The incidence of the disorder is one in 18,700, which was three times higher than the worldwide incidence.[9] Several unique mutations were found in western Hungary, and the study revealed that 10 out of 21 different mutations were unique to the Hungarian population.[56] The mutations identified in the Hungarian population in NBS program effectively helped to detect the patients with BTD deficiency. The mutation T532M is a common mutation among the Romanian Gypsy population.[57]
In India, a 7-year-old boy with recurrent myelopathy had a BTD deficiency. He had a lot of clinical significance, including progressive weakness in four limbs and difficulties in swallowing and breathing. He didn't have manifestation of hearing impairment, alopecia, and skin rashes. At the age of 5 years, he had a respiratory problem, but tomography of the chest and bronchoscopy showed negative results. His illness did not respond to steroids and immunotherapy such as intravenous immunoglobulin. A oral supplementation of biotin, thiamine, and multivitamins helped him recover from this illness in 1 month, after which medication was discontinued. At the age of 7 years, he was examined by various clinical analyses, but most of them were normal. However, the cerebrospinal fluid lactate level was slightly elevated, and the BTD level was significantly lower than normal. The molecular analysis revealed an H447Y homozygous mutation in his BTD gene, and he was not born to a consanguineous parent.[58] Therefore, NBS program is very important, which may help in early detection of this deficiency. In cases with the positive results, the biotin supplement helps to avoid those clinical signs and maintain the proper biotin metabolic cycle in the human body.
The homozygous mutation L215F is frequently found in the Northeastern region of Poland. There were three out of four patients affected by this L215F mutation, and the patients had symptoms such as hearing loss or vision problems.[59] In Italy, a patient showed a new variant of T492I with a combination of D444H, and the mother didn't have a similar mutation in her genomic DNA, which suggests the mutation is due to a de novo origin of maternal germline mosaicism.[60] The prevalence of BTD deficiency in the European population is estimated at one in 61,000.[61] A 10-year long NBS program of BTD deficiency revealed that 75 neonates were affected among 579,812 newborns in the regions of Tuscany and Umbria in Italy. The incidence is approximately one in 6,300 births, which is 10 times higher than worldwide.[62] In Italy, NBS by the Regional Screening Centre of Verona from 2014 until the end of 2020 found 49 BTD patients among 293,784 newborns screened. Among 49 BTD patients, five were affected with profound deficiency, and the other 44 were affected with partial BTD deficiency. They were treated with oral biotin supplement. The patients with profound deficiency were given a 10 mg/d dosage, and the partial BTD deficiency patients with 5 mg/d. None of them has shown clinical signs or symptoms, including patients with profound BTD deficiency during diagnosis and biotin treatment follow-up. The newborn screening program is a major help to those patients trying to prevent severe neurological and other problems. The mutation c.1330G > C (D444H) is found in all partial BTD patients, its pathogenic variant in compound heterozygosity. This mutation has a high prevalence among the European population and was also reported in other regions of the world.[63] These results indicate, that D444H is the common variant in BTD gene and that may be used as a biomarker for the purpose of prenatal diagnosis.
Analysis of BTD mutations in the Brazilian population, combined with the NBS, revealed that there were 119 neonates affected by BTD deficiency from June 2013 to December 2017 in Minas Gerais, Brazil. In 2013, BTD deficiency testing was made mandatory in NBS, which successfully helped detect people born with BTD deficiency.[24] Because of the higher rate of consanguineous marriages, Iran has a high prevalence of BTD deficiency, which affects the BTD homozygous mutations in the Iranian population.[64]
The BTD deficiency test is recommended for all newborns, especially if their sibling is affected with BTD deficiency, which helps prevent clinical symptoms. Several mutations of the BTD gene have been found in the Malaysian population. A study demonstrated screening of 1,434 patients, nine of whom were affected by BTD deficiency. One of the patients had been affected by encephalopathy and had several symptoms such as developmental delay, alopecia, and sparse eyebrows. In Malaysia, BTD deficiency screening is not mandatory for NBS, but neonates are at high risk of BTD deficiency.[65]
Senanayake et al discovered the novel exon 1 deletion in BTD gene in Sri Lankan child, the homozygous contiguous deletion of entire exon1 in BTD gene has failed to produce active BTD enzyme. Exon-1 is the starting point of the BTD enzyme and the leading signal sequence. The child also had a deletion in the HACL1 gene and an exon 1 deletion in the COLQ gene.[66] In Jordanian study, several mutations of the BTD gene have been identified, and the novel mutation V150E has been affected in one patient in the Jordanian population. In addition, the endogamy rate is higher in Jordanian and Middle Eastern countries, which has led to the incidence of autosomal inherited disorders. Early childhood or NBS helps to detect such a problem and is easily treated to avoid severe consequences.[67] Mutation Q456H has been found in the Austrian population. This mutation causing the profound deficiency is common in NBS in the United States.[26] BTD deficiency is rare among the Swedish population, but due to immigration and consanguineous marriages, some of them are affected with BTD deficiency. Newborn screening is mandatory in Sweden, which earlier detects the deficiency and treats them with biotin supplements. The BTD deficiency among the Swedish population demonstrates the disease's heterogeneity.[68]
Clinical Significance
Patients with BTD deficiency experience a variety of symptoms. The profound BTD patients are caused by severe symptoms such as neurological disorders, vision problems, cutaneous manifestations, and hearing loss. The hearing and vision loss and neurological problems are not reversible. The cutaneous symptoms are reversible when treated with oral supplementation. Epilepsy, alopecia, and seizures are common among BTD patients. Urinary organic acid levels are abnormal in BTD patients, such as propionic acid, lactic acid, alanine, and pyruvate acid. BTD deficiency also affects the patient's immunity, which declines cellular and humoral immunity. Sometimes children with BTD are caused by both Candida and bacterial infections.[69] Additionally, BTD deficiency can cause fetal malformation.[70]
Approximately, 76% of the symptomatic BTD patients are affected by sensory hearing loss. These hearing losses are not reversible by biotin supplementation. In the United States, a study on 33 children revealed that two-thirds of the children of profound symptomatic patients are affected by hearing loss.[7] Acrodermatitis dysmetabolic was found in BTD patients in India. After taking oral biotin supplements, the lesion disappeared in 2 weeks, and he was advised to take supplements lifelong.[71] In India, a patient has Ohtahara syndrome, which is associated with BTD deficiency. The syndrome is a rare epileptic encephalopathy condition.[72] Spastic paraparesis has rarely been reported in a patient with BTD deficiency, which is involved in the spinal cord.[73] [74]
Efficacy of Biotin Treatment
A symptomatic BTD patient was initially given a 20 mg biotin dose once a day, gradually reducing it to 5 mg based on the patient's clinical symptoms. In addition, asymptomatic BTD patients take 5 mg of oral biotin daily. A study of 22 Polish pediatric patients was screened for biotin follow-up for 20 years by Szymanska et al.[59] They routinely monitored the patient's urine organic acid (3-hydroxyisovaleric acid) by gas chromatography mass spectrometry. Once a year, ophthalmological, audiological, and neurological evaluations were also conducted. A rapid improvement in psychomotor skills was observed in most of the symptomatic patients after the initiation of biotin supplementation. The cutaneous symptoms of skin rash disappeared, and progressive optic nerve atrophy was observed before starting treatment. During the treatment, there was no further deterioration in the optic nerve. Sensorial hearing loss in all patients was not reversed, but no progression was observed.
Most profound BTD patients need to take oral biotin supplementation for life, which is the most effective and safest treatment. Partial patients are advised to take a low dose of oral biotin supplements.[75] Oral biotin supplementation at the pre-symptomatic stage helps to avoid the symptoms, including optic atrophy. The antiepileptic medication and some medications are not responding to the BTD patient.[66] For a child with severe BTD deficiency such as hyperammonemia or metabolic acidosis, it's necessary to limit protein, correct acidosis, and supplement glucose.[75] In India, a child with BTD deficiency has been treated with a biotin supplement, and his perioral lesions, alopecia, and seborrheic have disappeared after high doses of biotin treatment.[76] Intake of raw eggs must be avoided during the oral supplementation of biotin, which contains avidin that binds to the biotin. A follow-up study revealed that after taking a biotin supplement in two patients with encephalopathy, it was found that the cerebral volume had been reversed in image findings.[73] Newborn screening programs have the potential to detect BTD deficiency in all infants and provide early diagnosis. Pre-symptomatic treatments help prevent this disorder's consequences, particularly neurological problems, and hearing and vision loss.[77]
Conclusion
Worldwide, the estimated carrier frequency of the BTD mutation is approximately one in 120, and the combined incidence of profound and partial BTD deficiency cases is one in 60,000. The NBS programs are life-saving for patients with BTD deficiency. Developing and under developing countries do not have this system because of their economic conditions and insufficient hospitals. In the United States, Turkey, Hungary, Brazil, and European countries, screening of newborns for BTD and other disorders is mandatory, which helps to prevent or treat them earlier. Therefore, it is essential to implement the NBS program in developing and low-income countries to overcome the BTD deficiency, which is life-saving for newborns from severe disorders. In addition, there is a need for identification of biomarkers (molecular screening) for the BTD deficiency to provide a better treatment strategy. Treatment for partial BTD patients is still being debated because the biotin dosage and treatment period for this disease remain unclear. Moreover, for families having a history of BTD deficiency, molecular detection of the BTD gene in the father and mother during the pregnancy period could be helpful for the early detection of BTD deficiency in a newborn and provide a better treatment strategy.
Conflict of Interest
None declared.
Acknowledgments
All authors thank Saveetha Dental College and Hospitals for providing support. Author thanks Nausheen Raheema for helping in language editing for this manuscript.
Authors' Contribution
B.K. undertook literature mining from various reputed databases, drafted the manuscript, and prepared illustrations. P.A. gave the concept for this article and is responsible for manuscript proof reading and validated the entire manuscript. H.K.N. and V.P.J. corrected the final manuscript draft.
-
References
- 1 Hymes J, Wolf B. Biotinidase and its roles in biotin metabolism. Clin Chim Acta 1996; 255 (01) 1-11
- 2 Wolf B, Grier RE, Allen RJ, Goodman SI, Kien CL. Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta 1983; 131 (03) 273-281
- 3 Pispa J. Animal biotinidase. Ann Med Exp Biol Fenn 1965; 43: 5 , 1–39
- 4 Hymes J, Fleischhauer K, Wolf B. Biotinylation of histones by human serum biotinidase: assessment of biotinyl-transferase activity in sera from normal individuals and children with biotinidase deficiency. Biochem Mol Med 1995; 56 (01) 76-83
- 5 Li H, Spencer L, Nahhas F. et al. Novel mutations causing biotinidase deficiency in individuals identified by newborn screening in Michigan including an unique intronic mutation that alters mRNA expression of the biotinidase gene. Mol Genet Metab 2014; 112 (03) 242-246
- 6 Wiltink RC, Kruijshaar ME, van Minkelen R. et al. Neonatal screening for profound biotinidase deficiency in the Netherlands: consequences and considerations. Eur J Hum Genet 2016; 24 (10) 1424-1429
- 7 Wolf B, Jensen K, Hüner G. et al. Seventeen novel mutations that cause profound biotinidase deficiency. Mol Genet Metab 2002; 77 (1-2): 108-111
- 8 Wolf B, Grier RE, Secor McVoy JR, Heard GS. Biotinidase deficiency: a novel vitamin recycling defect. J Inherit Metab Dis 1985; 8 (Suppl. 01) 53-58
- 9 Wolf B, Heard GS, Weissbecker KA, McVoy JR, Grier RE, Leshner RT. Biotinidase deficiency: initial clinical features and rapid diagnosis. Ann Neurol 1985; 18 (05) 614-617
- 10 Wolf B, Grier RE, Allen RJ. et al. Phenotypic variation in biotinidase deficiency. J Pediatr 1983; 103 (02) 233-237
- 11 Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med 2012; 14 (06) 565-575
- 12 McVoy JR, Levy HL, Lawler M. et al. Partial biotinidase deficiency: clinical and biochemical features. J Pediatr 1990; 116 (01) 78-83
- 13 Burlina AB, Dermikol M, Mantau A. et al. Increased plasma biotinidase activity in patients with glycogen storage disease type Ia: effect of biotin supplementation. J Inherit Metab Dis 1996; 19 (02) 209-212
- 14 Hymes J, Wolf B. Human biotinidase isn't just for recycling biotin. J Nutr 1999; 129 (suppl 2S): 485S-489S
- 15 Wolf B. The neurology of biotinidase deficiency. Mol Genet Metab 2011; 104 (1-2): 27-34
- 16 Rybak LP, Whitworth C, Scott V, Weberg AD, Bhardwaj B. Rat as a potential model for hearing loss in biotinidase deficiency. Ann Otol Rhinol Laryngol 1991; 100 (4 Pt 1): 294-300
- 17 Micó SI, Jiménez RD, Salcedo EM, Martínez HA, Mira AP, Fernández CC. Epilepsy in biotinidase deficiency after biotin treatment. JIMD Rep 2012; 4: 75-78
- 18 Thodi G, Molou E, Georgiou V. et al. Mutational analysis for biotinidase deficiency of a Greek patients' cohort ascertained through expanded newborn screening. J Hum Genet 2011; 56 (12) 861-865
- 19 Heard GS, Secor McVoy JR, Wolf B. A screening method for biotinidase deficiency in newborns. Clin Chem 1984; 30 (01) 125-127
- 20 Broda E, Baumgartner ER, Scholl S. et al. Biotinidase determination in serum and dried blood spots--high sensitivity fluorimetric ultramicro-assay. Clin Chim Acta 2001; 314 (1-2): 175-185
- 21 Cole H, Reynolds TR, Lockyer JM. et al. Human serum biotinidase. cDNA cloning, sequence, and characterization. J Biol Chem 1994; 269 (09) 6566-6570
- 22 Knight HC, Reynolds TR, Meyers GA, Pomponio RJ, Buck GA, Wolf B. Structure of the human biotinidase gene. Mamm Genome 1998; 9 (04) 327-330
- 23 Swango KL, Hymes J, Brown P, Wolf B. Amino acid homologies between human biotinidase and bacterial aliphatic amidases: putative identification of the active site of biotinidase. Mol Genet Metab 2000; 69 (02) 111-115
- 24 Carvalho NO, Del Castillo DM, Januário JN. et al. Novel mutations causing biotinidase deficiency in individuals identified by the newborn screening program in Minas Gerais, Brazil. Am J Med Genet A 2019; 179 (06) 978-982
- 25 Iqbal F, Item CB, Vilaseca MA. et al. The identification of novel mutations in the biotinidase gene using denaturing high pressure liquid chromatography (dHPLC). Mol Genet Metab 2010; 100 (01) 42-45
- 26 Mühl A, Möslinger D, Item CB, Stöckler-Ipsiroglu S. Molecular characterisation of 34 patients with biotinidase deficiency ascertained by newborn screening and family investigation. Eur J Hum Genet 2001; 9 (04) 237-243
- 27 Ye J, Wang T, Han LS. et al. Diagnosis, treatment, follow-up and gene mutation analysis in four Chinese children with biotinidase deficiency. J Inherit Metab Dis 2009; 32 (Suppl. 01) S295-S302
- 28 Wolf B, Spencer R, Gleason T. Hearing loss is a common feature of symptomatic children with profound biotinidase deficiency. J Pediatr 2002; 140 (02) 242-246
- 29 Pindolia K, Jordan M, Wolf B. Analysis of mutations causing biotinidase deficiency. Hum Mutat 2010; 31 (09) 983-991
- 30 Norrgard KJ, Pomponio RJ, Hymes J, Wolf B. Mutations causing profound biotinidase deficiency in children ascertained by newborn screening in the United States occur at different frequencies than in symptomatic children. Pediatr Res 1999; 46 (01) 20-27
- 31 Procter M, Wolf B, Mao R. Forty-eight novel mutations causing biotinidase deficiency. Mol Genet Metab 2016; 117 (03) 369-372
- 32 Norrgard KJ, Pomponio RJ, Swango KL. et al. Mutation (Q456H) is the most common cause of profound biotinidase deficiency in children ascertained by newborn screening in the United States. Biochem Mol Med 1997; 61 (01) 22-27
- 33 Swango KL, Demirkol M, Hüner G. et al. Partial biotinidase deficiency is usually due to the D444H mutation in the biotinidase gene. Hum Genet 1998; 102 (05) 571-575
- 34 Cowan TM, Kazerouni NN, Dharajiya N. et al. Increased incidence of profound biotinidase deficiency among Hispanic newborns in California. Mol Genet Metab 2012; 106 (04) 485-487
- 35 Karaca M, Özgül RK, Ünal Ö. et al. Detection of biotinidase gene mutations in Turkish patients ascertained by newborn and family screening. Eur J Pediatr 2015; 174 (08) 1077-1084
- 36 Pindolia K, Jensen K, Wolf B. Three dimensional structure of human biotinidase: computer modeling and functional correlations. Mol Genet Metab 2007; 92 (1-2): 13-22
- 37 Sivri HS, Genç GA, Tokatli A. et al. Hearing loss in biotinidase deficiency: genotype-phenotype correlation. J Pediatr 2007; 150 (04) 439-442
- 38 Wolf B, Jensen KP, Barshop B. et al. Biotinidase deficiency: novel mutations and their biochemical and clinical correlates. Hum Mutat 2005; 25 (04) 413
- 39 Pace HC, Brenner C. The nitrilase superfamily: classification, structure and function. Genome Biol 2001; 2 (01) REVIEWS0001
- 40 Chedrawi AK, Ali A, Al Hassnan ZN, Faiyaz-Ul-Haque M, Wolf B. Profound biotinidase deficiency in a child with predominantly spinal cord disease. J Child Neurol 2008; 23 (09) 1043-1048
- 41 Kasapkara ÇS, Akar M, Özbek MN. et al. Mutations in BTD gene causing biotinidase deficiency: a regional report. J Pediatr Endocrinol Metab 2015; 28 (3-4): 421-424
- 42 Baykal T, Gokcay G, Gokdemir Y. et al. Asymptomatic adults and older siblings with biotinidase deficiency ascertained by family studies of index cases. J Inherit Metab Dis 2005; 28 (06) 903-912
- 43 Seker Yilmaz B, Mungan NO, Kor D. et al. Twenty-seven mutations with three novel pathogenic variants causing biotinidase deficiency: a report of 203 patients from the southeastern part of Turkey. J Pediatr Endocrinol Metab 2018; 31 (03) 339-343
- 44 Liu Z, Zhao X, Sheng H. et al. Clinical features, BTD gene mutations, and their functional studies of eight symptomatic patients with biotinidase deficiency from Southern China. Am J Med Genet A 2018; 176 (03) 589-596
- 45 Wiznitzer M, Bangert BA. Biotinidase deficiency: clinical and MRI findings consistent with myelopathy. Pediatr Neurol 2003; 29 (01) 56-58
- 46 Yang Y, Li C, Qi Z. et al. Spinal cord demyelination associated with biotinidase deficiency in 3 Chinese patients. J Child Neurol 2007; 22 (02) 156-160
- 47 Pomponio RJ, Coskun T, Demirkol M. et al. Novel mutations cause biotinidase deficiency in Turkish children. J Inherit Metab Dis 2000; 23 (02) 120-128
- 48 Yang Y, Yang JY, Chen XJ. Biotinidase deficiency characterized by skin and hair findings. Clin Dermatol 2020; 38 (04) 477-483
- 49 Geng J, Sun Y, Zhao Y. et al. Two novel BTD mutations causing profound biotinidase deficiency in a Chinese patient. Mol Genet Genomic Med 2021; 9 (02) e1591
- 50 Karimzadeh P, Ahmadabadi F, Jafari N. et al. Biotinidase deficiency: a reversible neurometabolic disorder (an Iranian pediatric case series). Iran J Child Neurol 2013; 7 (04) 47-52
- 51 Singh A, Lomash A, Pandey S, Kapoor S. Clinical, biochemical and outcome profile of biotinidase deficient patients from tertiary centre in Northern India. J Clin Diagn Res 2015; 9 (12) SC08-SC10
- 52 Sarafoglou K, Bentler K, Gaviglio A. et al. High incidence of profound biotinidase deficiency detected in newborn screening blood spots in the Somalian population in Minnesota. J Inherit Metab Dis 2009; 32 (Suppl. 01) S169-S173
- 53 Norrgard KJ, Pomponio RJ, Swango KL. et al. Double mutation (A171T and D444H) is a common cause of profound biotinidase deficiency in children ascertained by newborn screening the United States. Mutations in brief no. 128. Online. Hum Mutat 1998; 11 (05) 410
- 54 Pomponio RJ, Hymes J, Reynolds TR. et al. Mutations in the human biotinidase gene that cause profound biotinidase deficiency in symptomatic children: molecular, biochemical, and clinical analysis. Pediatr Res 1997; 42 (06) 840-848
- 55 Milánkovics I, Németh K, Somogyi C, Schuler A, Fekete G. High frequencies of biotinidase (BTD) gene mutations in the Hungarian population. J Inherit Metab Dis 2010; 33 (Suppl. 03) S289-S292
- 56 Milánkovics I, Kámory E, Csókay B, Fodor F, Somogyi C, Schuler A. Mutations causing biotinidase deficiency in children ascertained by newborn screening in Western Hungary. Mol Genet Metab 2007; 90 (03) 345-348
- 57 László A, Schuler EA, Sallay E. et al. Neonatal screening for biotinidase deficiency in Hungary: clinical, biochemical and molecular studies. J Inherit Metab Dis 2003; 26 (07) 693-698
- 58 Raha S, Udani V. Biotinidase deficiency presenting as recurrent myelopathy in a 7-year-old boy and a review of the literature. Pediatr Neurol 2011; 45 (04) 261-264
- 59 Szymańska E, Średzińska M, Ługowska A, Pajdowska M, Rokicki D, Tylki-Szymańska A. Outcomes of oral biotin treatment in patients with biotinidase deficiency - twenty years follow-up. Mol Genet Metab Rep 2015; 5: 33-35
- 60 Tonin R, Caciotti A, Funghini S. et al. Biotinidase deficiency due to a de novo mutation or gonadal mosaicism in a first child. Clin Chim Acta 2015; 445: 70-72
- 61 Weber P, Scholl S, Baumgartner ER. Outcome in patients with profound biotinidase deficiency: relevance of newborn screening. Dev Med Child Neurol 2004; 46 (07) 481-484
- 62 Funghini S, Tonin R, Malvagia S. et al. High frequency of biotinidase deficiency in Italian population identified by newborn screening. Mol Genet Metab Rep 2020; 25: 100689
- 63 Maguolo A, Rodella G, Dianin A. et al. Newborn screening for biotinidase deficiency. The experience of a regional center in Italy. Front Pediatr 2021; 9: 661416
- 64 Torkamandi S, Rezaei S, Mirfakhraie R, Golmohamadi S, Gholami M. The novel homozygous p.Asn197_Ser201del mutation in BTD gene is associated with profound biotinidase deficiency in an Iranian consanguineous family. Mol Biol Rep 2020; 47 (05) 4021-4027
- 65 Mardhiah M, Azize NAA, Yakob Y. et al. Clinical, biochemical and mutational findings in biotinidase deficiency among Malaysian population. Mol Genet Metab Rep 2019; 22: 100548
- 66 Senanayake DN, Jasinge EA, Pindolia K. et al. First contiguous gene deletion causing biotinidase deficiency: the enzyme deficiency in three Sri Lankan children. Mol Genet Metab Rep 2015; 2: 81-84
- 67 Al-Eitan LN, Alqa'qa' K, Amayreh W. et al. Identification and characterization of BTD gene mutations in Jordanian children with biotinidase deficiency. J Pers Med 2020; 10 (01) 4
- 68 Ohlsson A, Guthenberg C, Holme E, von Döbeln U. Profound biotinidase deficiency: a rare disease among native Swedes. J Inherit Metab Dis 2010; 33 (Suppl. 03) S175-S180
- 69 Yanling Y. Biotin and Biotinase Deficiency. Basic and Clinical Diseases of Pediatric Nervous System. 2nd ed. Beijing: People's Health Publishing House; 2009: 634-635
- 70 Takechi R, Taniguchi A, Ebara S, Fukui T, Watanabe T. Biotin deficiency affects the proliferation of human embryonic palatal mesenchymal cells in culture. J Nutr 2008; 138 (04) 680-684
- 71 Patra S, Senthilnathan G, Bhari N. Acrodermatitis enteropathica-like skin eruption with neonatal seizures in a child with biotinidase deficiency. Clin Exp Dermatol 2020; 45 (02) 266-267
- 72 Singhi P, Ray M. Ohtahara syndrome with biotinidase deficiency. J Child Neurol 2011; 26 (04) 507-509
- 73 Desai S, Ganesan K, Hegde A. Biotinidase deficiency: a reversible metabolic encephalopathy. Neuroimaging and MR spectroscopic findings in a series of four patients. Pediatr Radiol 2008; 38 (08) 848-856
- 74 Honavar M, Janota I, Neville BG, Chalmers RA. Neuropathology of biotinidase deficiency. Acta Neuropathol 1992; 84 (04) 461-464
- 75 Wolf B. Disorders of biotin metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D. eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001: 3935-3962
- 76 Mukhopadhyay D, Das MK, Dhar S, Mukhopadhyay M. Multiple carboxylase deficiency (late onset) due to deficiency of biotinidase. Indian J Dermatol 2014; 59 (05) 502-504
- 77 Möslinger D, Mühl A, Suormala T, Baumgartner R, Stöckler-Ipsiroglu S. Molecular characterisation and neuropsychological outcome of 21 patients with profound biotinidase deficiency detected by newborn screening and family studies. Eur J Pediatr 2003; 162 (Suppl. 01) S46-S49
- 78 Neto EC, Schulte J, Rubim R. et al. Newborn screening for biotinidase deficiency in Brazil: biochemical and molecular characterizations. Brazilian journal of medical and biological research. Rev Bras Pesqui Med Biol 2004; 37 (03) 295-299
- 79 Canda E, Yazici H, Er E. et al. Single center experience of biotinidase deficiency: 259 patients and six novel mutations. J Pediatr Endocrinol Metab 2018; 31 (08) 917-926
- 80 Couce Pico ML, Martinón-Torres F, Castiñeiras DE, Alonso-Fernández JR, Fraga JM. Deficiencia de biotinidasa: importancia de su diagnóstico neonatal y tratamiento precoz. [Biotinidase deficiency: importance of its neonatal diagnosis and early treatment] An Esp Pediatr 1999; 50 (05) 504-506
- 81 Hsu RH, Chien YH, Hwu WL. et al. Genotypic and phenotypic correlations of biotinidase deficiency in the Chinese population. Orphanet J Rare Dis 2019; 14 (01) 6
- 82 Murry JB, Machini K, Ceyhan-Birsoy O. et al; BabySeq Project Team. Reconciling newborn screening and a novel splice variant in BTD associated with partial biotinidase deficiency: a BabySeq Project case report. Cold Spring Harb Mol Case Stud 2018; 4 (04) a002873
Address for correspondence
Publication History
Received: 24 January 2022
Accepted: 06 September 2022
Article published online:
01 November 2022
© 2022. Thieme. All rights reserved.
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Hymes J, Wolf B. Biotinidase and its roles in biotin metabolism. Clin Chim Acta 1996; 255 (01) 1-11
- 2 Wolf B, Grier RE, Allen RJ, Goodman SI, Kien CL. Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta 1983; 131 (03) 273-281
- 3 Pispa J. Animal biotinidase. Ann Med Exp Biol Fenn 1965; 43: 5 , 1–39
- 4 Hymes J, Fleischhauer K, Wolf B. Biotinylation of histones by human serum biotinidase: assessment of biotinyl-transferase activity in sera from normal individuals and children with biotinidase deficiency. Biochem Mol Med 1995; 56 (01) 76-83
- 5 Li H, Spencer L, Nahhas F. et al. Novel mutations causing biotinidase deficiency in individuals identified by newborn screening in Michigan including an unique intronic mutation that alters mRNA expression of the biotinidase gene. Mol Genet Metab 2014; 112 (03) 242-246
- 6 Wiltink RC, Kruijshaar ME, van Minkelen R. et al. Neonatal screening for profound biotinidase deficiency in the Netherlands: consequences and considerations. Eur J Hum Genet 2016; 24 (10) 1424-1429
- 7 Wolf B, Jensen K, Hüner G. et al. Seventeen novel mutations that cause profound biotinidase deficiency. Mol Genet Metab 2002; 77 (1-2): 108-111
- 8 Wolf B, Grier RE, Secor McVoy JR, Heard GS. Biotinidase deficiency: a novel vitamin recycling defect. J Inherit Metab Dis 1985; 8 (Suppl. 01) 53-58
- 9 Wolf B, Heard GS, Weissbecker KA, McVoy JR, Grier RE, Leshner RT. Biotinidase deficiency: initial clinical features and rapid diagnosis. Ann Neurol 1985; 18 (05) 614-617
- 10 Wolf B, Grier RE, Allen RJ. et al. Phenotypic variation in biotinidase deficiency. J Pediatr 1983; 103 (02) 233-237
- 11 Wolf B. Biotinidase deficiency: “if you have to have an inherited metabolic disease, this is the one to have”. Genet Med 2012; 14 (06) 565-575
- 12 McVoy JR, Levy HL, Lawler M. et al. Partial biotinidase deficiency: clinical and biochemical features. J Pediatr 1990; 116 (01) 78-83
- 13 Burlina AB, Dermikol M, Mantau A. et al. Increased plasma biotinidase activity in patients with glycogen storage disease type Ia: effect of biotin supplementation. J Inherit Metab Dis 1996; 19 (02) 209-212
- 14 Hymes J, Wolf B. Human biotinidase isn't just for recycling biotin. J Nutr 1999; 129 (suppl 2S): 485S-489S
- 15 Wolf B. The neurology of biotinidase deficiency. Mol Genet Metab 2011; 104 (1-2): 27-34
- 16 Rybak LP, Whitworth C, Scott V, Weberg AD, Bhardwaj B. Rat as a potential model for hearing loss in biotinidase deficiency. Ann Otol Rhinol Laryngol 1991; 100 (4 Pt 1): 294-300
- 17 Micó SI, Jiménez RD, Salcedo EM, Martínez HA, Mira AP, Fernández CC. Epilepsy in biotinidase deficiency after biotin treatment. JIMD Rep 2012; 4: 75-78
- 18 Thodi G, Molou E, Georgiou V. et al. Mutational analysis for biotinidase deficiency of a Greek patients' cohort ascertained through expanded newborn screening. J Hum Genet 2011; 56 (12) 861-865
- 19 Heard GS, Secor McVoy JR, Wolf B. A screening method for biotinidase deficiency in newborns. Clin Chem 1984; 30 (01) 125-127
- 20 Broda E, Baumgartner ER, Scholl S. et al. Biotinidase determination in serum and dried blood spots--high sensitivity fluorimetric ultramicro-assay. Clin Chim Acta 2001; 314 (1-2): 175-185
- 21 Cole H, Reynolds TR, Lockyer JM. et al. Human serum biotinidase. cDNA cloning, sequence, and characterization. J Biol Chem 1994; 269 (09) 6566-6570
- 22 Knight HC, Reynolds TR, Meyers GA, Pomponio RJ, Buck GA, Wolf B. Structure of the human biotinidase gene. Mamm Genome 1998; 9 (04) 327-330
- 23 Swango KL, Hymes J, Brown P, Wolf B. Amino acid homologies between human biotinidase and bacterial aliphatic amidases: putative identification of the active site of biotinidase. Mol Genet Metab 2000; 69 (02) 111-115
- 24 Carvalho NO, Del Castillo DM, Januário JN. et al. Novel mutations causing biotinidase deficiency in individuals identified by the newborn screening program in Minas Gerais, Brazil. Am J Med Genet A 2019; 179 (06) 978-982
- 25 Iqbal F, Item CB, Vilaseca MA. et al. The identification of novel mutations in the biotinidase gene using denaturing high pressure liquid chromatography (dHPLC). Mol Genet Metab 2010; 100 (01) 42-45
- 26 Mühl A, Möslinger D, Item CB, Stöckler-Ipsiroglu S. Molecular characterisation of 34 patients with biotinidase deficiency ascertained by newborn screening and family investigation. Eur J Hum Genet 2001; 9 (04) 237-243
- 27 Ye J, Wang T, Han LS. et al. Diagnosis, treatment, follow-up and gene mutation analysis in four Chinese children with biotinidase deficiency. J Inherit Metab Dis 2009; 32 (Suppl. 01) S295-S302
- 28 Wolf B, Spencer R, Gleason T. Hearing loss is a common feature of symptomatic children with profound biotinidase deficiency. J Pediatr 2002; 140 (02) 242-246
- 29 Pindolia K, Jordan M, Wolf B. Analysis of mutations causing biotinidase deficiency. Hum Mutat 2010; 31 (09) 983-991
- 30 Norrgard KJ, Pomponio RJ, Hymes J, Wolf B. Mutations causing profound biotinidase deficiency in children ascertained by newborn screening in the United States occur at different frequencies than in symptomatic children. Pediatr Res 1999; 46 (01) 20-27
- 31 Procter M, Wolf B, Mao R. Forty-eight novel mutations causing biotinidase deficiency. Mol Genet Metab 2016; 117 (03) 369-372
- 32 Norrgard KJ, Pomponio RJ, Swango KL. et al. Mutation (Q456H) is the most common cause of profound biotinidase deficiency in children ascertained by newborn screening in the United States. Biochem Mol Med 1997; 61 (01) 22-27
- 33 Swango KL, Demirkol M, Hüner G. et al. Partial biotinidase deficiency is usually due to the D444H mutation in the biotinidase gene. Hum Genet 1998; 102 (05) 571-575
- 34 Cowan TM, Kazerouni NN, Dharajiya N. et al. Increased incidence of profound biotinidase deficiency among Hispanic newborns in California. Mol Genet Metab 2012; 106 (04) 485-487
- 35 Karaca M, Özgül RK, Ünal Ö. et al. Detection of biotinidase gene mutations in Turkish patients ascertained by newborn and family screening. Eur J Pediatr 2015; 174 (08) 1077-1084
- 36 Pindolia K, Jensen K, Wolf B. Three dimensional structure of human biotinidase: computer modeling and functional correlations. Mol Genet Metab 2007; 92 (1-2): 13-22
- 37 Sivri HS, Genç GA, Tokatli A. et al. Hearing loss in biotinidase deficiency: genotype-phenotype correlation. J Pediatr 2007; 150 (04) 439-442
- 38 Wolf B, Jensen KP, Barshop B. et al. Biotinidase deficiency: novel mutations and their biochemical and clinical correlates. Hum Mutat 2005; 25 (04) 413
- 39 Pace HC, Brenner C. The nitrilase superfamily: classification, structure and function. Genome Biol 2001; 2 (01) REVIEWS0001
- 40 Chedrawi AK, Ali A, Al Hassnan ZN, Faiyaz-Ul-Haque M, Wolf B. Profound biotinidase deficiency in a child with predominantly spinal cord disease. J Child Neurol 2008; 23 (09) 1043-1048
- 41 Kasapkara ÇS, Akar M, Özbek MN. et al. Mutations in BTD gene causing biotinidase deficiency: a regional report. J Pediatr Endocrinol Metab 2015; 28 (3-4): 421-424
- 42 Baykal T, Gokcay G, Gokdemir Y. et al. Asymptomatic adults and older siblings with biotinidase deficiency ascertained by family studies of index cases. J Inherit Metab Dis 2005; 28 (06) 903-912
- 43 Seker Yilmaz B, Mungan NO, Kor D. et al. Twenty-seven mutations with three novel pathogenic variants causing biotinidase deficiency: a report of 203 patients from the southeastern part of Turkey. J Pediatr Endocrinol Metab 2018; 31 (03) 339-343
- 44 Liu Z, Zhao X, Sheng H. et al. Clinical features, BTD gene mutations, and their functional studies of eight symptomatic patients with biotinidase deficiency from Southern China. Am J Med Genet A 2018; 176 (03) 589-596
- 45 Wiznitzer M, Bangert BA. Biotinidase deficiency: clinical and MRI findings consistent with myelopathy. Pediatr Neurol 2003; 29 (01) 56-58
- 46 Yang Y, Li C, Qi Z. et al. Spinal cord demyelination associated with biotinidase deficiency in 3 Chinese patients. J Child Neurol 2007; 22 (02) 156-160
- 47 Pomponio RJ, Coskun T, Demirkol M. et al. Novel mutations cause biotinidase deficiency in Turkish children. J Inherit Metab Dis 2000; 23 (02) 120-128
- 48 Yang Y, Yang JY, Chen XJ. Biotinidase deficiency characterized by skin and hair findings. Clin Dermatol 2020; 38 (04) 477-483
- 49 Geng J, Sun Y, Zhao Y. et al. Two novel BTD mutations causing profound biotinidase deficiency in a Chinese patient. Mol Genet Genomic Med 2021; 9 (02) e1591
- 50 Karimzadeh P, Ahmadabadi F, Jafari N. et al. Biotinidase deficiency: a reversible neurometabolic disorder (an Iranian pediatric case series). Iran J Child Neurol 2013; 7 (04) 47-52
- 51 Singh A, Lomash A, Pandey S, Kapoor S. Clinical, biochemical and outcome profile of biotinidase deficient patients from tertiary centre in Northern India. J Clin Diagn Res 2015; 9 (12) SC08-SC10
- 52 Sarafoglou K, Bentler K, Gaviglio A. et al. High incidence of profound biotinidase deficiency detected in newborn screening blood spots in the Somalian population in Minnesota. J Inherit Metab Dis 2009; 32 (Suppl. 01) S169-S173
- 53 Norrgard KJ, Pomponio RJ, Swango KL. et al. Double mutation (A171T and D444H) is a common cause of profound biotinidase deficiency in children ascertained by newborn screening the United States. Mutations in brief no. 128. Online. Hum Mutat 1998; 11 (05) 410
- 54 Pomponio RJ, Hymes J, Reynolds TR. et al. Mutations in the human biotinidase gene that cause profound biotinidase deficiency in symptomatic children: molecular, biochemical, and clinical analysis. Pediatr Res 1997; 42 (06) 840-848
- 55 Milánkovics I, Németh K, Somogyi C, Schuler A, Fekete G. High frequencies of biotinidase (BTD) gene mutations in the Hungarian population. J Inherit Metab Dis 2010; 33 (Suppl. 03) S289-S292
- 56 Milánkovics I, Kámory E, Csókay B, Fodor F, Somogyi C, Schuler A. Mutations causing biotinidase deficiency in children ascertained by newborn screening in Western Hungary. Mol Genet Metab 2007; 90 (03) 345-348
- 57 László A, Schuler EA, Sallay E. et al. Neonatal screening for biotinidase deficiency in Hungary: clinical, biochemical and molecular studies. J Inherit Metab Dis 2003; 26 (07) 693-698
- 58 Raha S, Udani V. Biotinidase deficiency presenting as recurrent myelopathy in a 7-year-old boy and a review of the literature. Pediatr Neurol 2011; 45 (04) 261-264
- 59 Szymańska E, Średzińska M, Ługowska A, Pajdowska M, Rokicki D, Tylki-Szymańska A. Outcomes of oral biotin treatment in patients with biotinidase deficiency - twenty years follow-up. Mol Genet Metab Rep 2015; 5: 33-35
- 60 Tonin R, Caciotti A, Funghini S. et al. Biotinidase deficiency due to a de novo mutation or gonadal mosaicism in a first child. Clin Chim Acta 2015; 445: 70-72
- 61 Weber P, Scholl S, Baumgartner ER. Outcome in patients with profound biotinidase deficiency: relevance of newborn screening. Dev Med Child Neurol 2004; 46 (07) 481-484
- 62 Funghini S, Tonin R, Malvagia S. et al. High frequency of biotinidase deficiency in Italian population identified by newborn screening. Mol Genet Metab Rep 2020; 25: 100689
- 63 Maguolo A, Rodella G, Dianin A. et al. Newborn screening for biotinidase deficiency. The experience of a regional center in Italy. Front Pediatr 2021; 9: 661416
- 64 Torkamandi S, Rezaei S, Mirfakhraie R, Golmohamadi S, Gholami M. The novel homozygous p.Asn197_Ser201del mutation in BTD gene is associated with profound biotinidase deficiency in an Iranian consanguineous family. Mol Biol Rep 2020; 47 (05) 4021-4027
- 65 Mardhiah M, Azize NAA, Yakob Y. et al. Clinical, biochemical and mutational findings in biotinidase deficiency among Malaysian population. Mol Genet Metab Rep 2019; 22: 100548
- 66 Senanayake DN, Jasinge EA, Pindolia K. et al. First contiguous gene deletion causing biotinidase deficiency: the enzyme deficiency in three Sri Lankan children. Mol Genet Metab Rep 2015; 2: 81-84
- 67 Al-Eitan LN, Alqa'qa' K, Amayreh W. et al. Identification and characterization of BTD gene mutations in Jordanian children with biotinidase deficiency. J Pers Med 2020; 10 (01) 4
- 68 Ohlsson A, Guthenberg C, Holme E, von Döbeln U. Profound biotinidase deficiency: a rare disease among native Swedes. J Inherit Metab Dis 2010; 33 (Suppl. 03) S175-S180
- 69 Yanling Y. Biotin and Biotinase Deficiency. Basic and Clinical Diseases of Pediatric Nervous System. 2nd ed. Beijing: People's Health Publishing House; 2009: 634-635
- 70 Takechi R, Taniguchi A, Ebara S, Fukui T, Watanabe T. Biotin deficiency affects the proliferation of human embryonic palatal mesenchymal cells in culture. J Nutr 2008; 138 (04) 680-684
- 71 Patra S, Senthilnathan G, Bhari N. Acrodermatitis enteropathica-like skin eruption with neonatal seizures in a child with biotinidase deficiency. Clin Exp Dermatol 2020; 45 (02) 266-267
- 72 Singhi P, Ray M. Ohtahara syndrome with biotinidase deficiency. J Child Neurol 2011; 26 (04) 507-509
- 73 Desai S, Ganesan K, Hegde A. Biotinidase deficiency: a reversible metabolic encephalopathy. Neuroimaging and MR spectroscopic findings in a series of four patients. Pediatr Radiol 2008; 38 (08) 848-856
- 74 Honavar M, Janota I, Neville BG, Chalmers RA. Neuropathology of biotinidase deficiency. Acta Neuropathol 1992; 84 (04) 461-464
- 75 Wolf B. Disorders of biotin metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D. eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001: 3935-3962
- 76 Mukhopadhyay D, Das MK, Dhar S, Mukhopadhyay M. Multiple carboxylase deficiency (late onset) due to deficiency of biotinidase. Indian J Dermatol 2014; 59 (05) 502-504
- 77 Möslinger D, Mühl A, Suormala T, Baumgartner R, Stöckler-Ipsiroglu S. Molecular characterisation and neuropsychological outcome of 21 patients with profound biotinidase deficiency detected by newborn screening and family studies. Eur J Pediatr 2003; 162 (Suppl. 01) S46-S49
- 78 Neto EC, Schulte J, Rubim R. et al. Newborn screening for biotinidase deficiency in Brazil: biochemical and molecular characterizations. Brazilian journal of medical and biological research. Rev Bras Pesqui Med Biol 2004; 37 (03) 295-299
- 79 Canda E, Yazici H, Er E. et al. Single center experience of biotinidase deficiency: 259 patients and six novel mutations. J Pediatr Endocrinol Metab 2018; 31 (08) 917-926
- 80 Couce Pico ML, Martinón-Torres F, Castiñeiras DE, Alonso-Fernández JR, Fraga JM. Deficiencia de biotinidasa: importancia de su diagnóstico neonatal y tratamiento precoz. [Biotinidase deficiency: importance of its neonatal diagnosis and early treatment] An Esp Pediatr 1999; 50 (05) 504-506
- 81 Hsu RH, Chien YH, Hwu WL. et al. Genotypic and phenotypic correlations of biotinidase deficiency in the Chinese population. Orphanet J Rare Dis 2019; 14 (01) 6
- 82 Murry JB, Machini K, Ceyhan-Birsoy O. et al; BabySeq Project Team. Reconciling newborn screening and a novel splice variant in BTD associated with partial biotinidase deficiency: a BabySeq Project case report. Cold Spring Harb Mol Case Stud 2018; 4 (04) a002873