Schlüsselwörter
Immuntherapie - Tumormilieu - monoklonale Antikörper - adoptive Zelltherapie
1. Einleitung
Das Gebiet der Immuntherapie war gerade in den letzten Jahren von signifikanten Fortschritten
geprägt, obwohl die Idee, das körpereigene Immunsystem für den Kampf gegen Krebs zu
verwenden, nicht neu ist. Denn bereits 1891 stimulierte William Coley das Immunsystem
von Sarkompatienten mit bakteriellen Bestandteilen und erzielte in einem Teil der
Patienten eine kurzzeitige Tumorreduktion [1]. Weshalb also setzte sich dieser initiale, immuntherapeutische Ansatz zunächst nicht
durch? Die Gründe hierfür sind vielfältig: Das Immunsystem ist ein hochkomplex reguliertes
und balanciertes System, welches durch stimulierende und inhibierende Komponenten
einerseits hochspezifisch auf Pathogene reagieren kann, andererseits eine überschießende
Reaktion verhindert und somit nicht den eigenen Körper angreift. Zudem sind Tumoren
sehr heterogen, da sie individuell entstehen und ihre Charakteristik vom Patienten
und dessen Ursprungsgewebe abhängt. Erschwerend kommt hinzu, dass das Ursprungsgewebe
der Tumoren eben nicht körperfremd ist, und somit wichtige Mechanismen der Immunantwort,
wie sie in der Erkennung von körperfremden Pathogenen funktionieren, nicht greifen.
Da es sich bei Coley um eine unspezifische, nicht gegen Tumorantigene gerichtete Reaktion
handelte, war der therapeutische Effekt nur transient. Diese aufgeführten Aspekte
begründen die anfänglichen Schwierigkeiten und enttäuschenden Ergebnisse, mit denen
sich die onkologische Immuntherapie auseinandersetzen musste und weiterhin muss. Doch
was hat sich zuletzt verändert? Warum werden aktuell in kaum einem anderen Bereich
so viele Mittel und Anstrengungen in die Entwicklung neuer Therapiemodalitäten investiert
wie im Bereich der Tumorimmunologie? Ein entscheidender Schritt war sicherlich die
Möglichkeit, mit neuen monoklonalen Antikörpern (mAb, engl. monoclonal Antibody) spezifisch
auf molekularer Ebene in das Tumorgeschehen einzugreifen. Nachdem über viele Jahre
versucht wurde, Immuntherapie im Sinne einer Immunaktivierung zu betreiben, hat man
seit einigen Jahren erkannt, dass von noch größerer Bedeutung die Antagonisierung
bzw. Beeinflussung von immunologischen Blockaden, „Kontrollpunkten“ (Checkpoints)
und immunsuppressiven Mechanismen ist. Dies wurde erstmals beim Malignen Melanom durch
Cytotoxic T-Lymphozyten-Associated Protein 4 (CTLA4)- [2] und Programmed Cell Death 1 (PD1)-spezifische [3] Antikörper erreicht. Die Ergebnisse waren derart überzeugend, dass Science diese Form der Immuntherapie als Durchbruch des Jahres adelte [4]. Zusätzlich besteht durch den wissenschaftlichen Fortschritt die Möglichkeit, körpereigene
Immunkomponenten auf spezifische (tumorale) Antigene „scharf zu stellen“, wie es bspw.
beim adoptiven T-Zelltransfer oder im Rahmen von Vakzinierungen geschieht. Viele dieser
Methoden sind aktuell und innovativ, stehen aber auch noch am Beginn ihrer (weiteren)
Entwicklung. Im Folgenden werden Chancen und Risiken der Immuntherapie beleuchtet.
Hierfür werden zunächst immunologische Grundlagen der Interaktion von Tumoren mit
dem Immunsystem erläutert, um darauf aufbauend unterschiedliche Therapieansätze vorzustellen.
Dies schließt sowohl einen Überblick über schon existierende Therapiemodalitäten,
als auch einen Ausblick auf zukünftige Entwicklungen mit ein.
2. Tumorimmunologische Grundlagen
2. Tumorimmunologische Grundlagen
Historisch und funktionell lässt sich das Immunsystem in zwei Arme unterteilen: Die
angeborene (native) Immunität bildet die erste Front der Immunabwehr und erkennt,
bekämpft und beseitigt – meist erfolgreich – fremde Pathogene schnell und effizient.
Allerdings ist die angeborene Immunität weder Antigen-spezifisch, noch lernfähig.
Das wiederum sind Eigenschaften der erworbenen (adaptiven) Immunität. Sie passt sich
spezifischen Antigenen an und kann dadurch eine langanhaltende, spezifisch angepasste
Immunantwort generieren. Beide Arme sind nicht autonom, sondern interagieren intensiv.
Zusätzlich wird immer deutlicher, dass die Grenzen zwischen angeborenem und adaptivem
Immunsystem fließend sind.
2.1. Angeborene Immunantwort
Zur angeborenen Immunantwort gehören physiologische Barrieren sowie humorale und zelluläre
Komponenten. Die zellulären Bestandteile zeichnen sich v. a. durch ihre Fähigkeit
aus, in Gewebe migrieren zu können und dort die Immunreaktion zu initiieren und gleichzeitig
weitere Komponenten des Immunsystems anzulocken. Dafür besitzen die meisten Zellen
der angeborenen Immunantwort die Fähigkeit zur Phagozytose, d. h. sie nehmen aktiv
Pathogene auf, verarbeiten sie weiter und präsentieren – je nach Zelltyp – Teile davon
auf ihrer Oberfläche auf Molekülen des Haupthistokompatibilitätskomplexes II (MHC
II, engl. Major Histocompatibility Complex II, [Abb. 1]). Zu den zellulären Komponenten der angeborenen Immunantwort zählen die Granulozyten,
die Makrophagen, die Dendritischen Zellen (DC) und die natürlichen Killerzellen (NK-Zellen).
Makrophagen können über Phagozytose aufgenommene Antigene weiterverarbeiten und effizient
über MHC II anderen Zellen präsentieren, um so eine Antigen-spezifische Antwort auszulösen.
Sie bilden daher eine wichtige Schnittstelle zwischen der angeborenen und der adaptiven
Immunantwort. In Abhängigkeit vom Umgebungsmilieu unterscheidet man bei den Makrophagen
im Wesentlichen 2 Phänotypen, den M1-und den M2-Phänotyp. Während der M1-Phänotyp
typischerweise durch Interferon Y (IFNγ) oder Bestandteile von Bakterien, wie Lipopolysaccharide
(LPS), aktiviert wird, resultiert der M2-Phänotyp überwiegend aus einer Stimulation
durch das antiinflammatorisch wirkende Interleukin (IL) 4. M1-Makrophagen werden durch
IFNγ, LPS, GM-CSF und TNF polarisiert, produzieren hauptsächlich pro-inflammatorische
Zytokine wie IL1, IL6, IL12, IL23 und TNFα und bewirken so eine T-Helferzellantwort,
die antitumoral gerichtet ist (TH1-Antwort). M2-Makrophagen werden v. a. durch IL4, IL10 und IL13 polarisiert, produzieren
selbst viel IL10 und TGFβ, aber wenig IL1, IL6, IL12 und TNF und bewirken daher eine
TH1-Suppression, TH2-Aktivierung sowie Immunsuppression und fördern die Wundheilung und Geweberegeneration.
Wegen immunsuppressiver Einflüsse im Tumormilieu polarisieren Tumor-assoziierte Makrophagen
(TAM) meist in Richtung des M2-Phänotyps [5]. Ihre Zahl im Tumor korreliert häufig mit Angiogenese, der Bildung von Metastasen
und der Tumorprogression [6]. Natürliche Killerzellen (NK-Zellen) erkennen pathologisch veränderte Zellen und
können sie direkt lysieren. Hierfür besitzen sie verschiedene Rezeptoren wie NKG2
(Natürliche Killer Gruppe 2) und KIR (Killerzell Immunoglobulin-ähnliche Rezeptoren).
Diese interagieren mit Liganden auf der Tumorzelle und senden stimulierende oder inhibitorische
Signale. NK-Zellen müssen selbst nicht aktiviert werden, ihre Aktivität lässt sich
jedoch durch Zytokine wie IL12, IFNα und IFNβ weiter steigern. NK-Zellen produzieren
selbst IFNγ und bewirken damit eine direkte Stimulation der an der Tumorabwehr beteiligten
Komponenten im Tumormilieu. Auch wenn die angeborene Immunantwort insbesondere in
der Fremderkennung eine wesentliche Rolle spielt, ist ihre Rolle – gerade die der
zellulären Bestandteile – in der Tumorentstehung und im Tumorprogress immer stärker
im Fokus der Forschung.
 Abb. 1 MHC-Moleküle. MHC-Moleküle sind auf kernhaltigen, körpereigenen Zellen (MHC I) und
Antigen-präsentierenden Zellen (MHC II) exprimiert. MHC I-Moleküle bestehen aus einer
α- und einer β-Untereinheit. Die α-Untereinheit enthält drei Domänen, von denen die
Domänen α1 und α2 die Antigenpräsentation übernehmen, und die α3 Domäne die Verankerung in der Zellmembran sichert. Das β2-Mikroglobulin stellt die vierte lösliche Domäne der MHC I-Moleküle dar. MHC II-Moleküle
bestehen aus 2 Untereinheiten, die beide in der Zellmembran verankert sind. Eine Untereinheit
(α- oder β) besteht aus je 2 Domänen, α1 und α2 bzw. β1 und β2, wobei die α1 und die β1 Domäne die Aufgabe der Antigenpräsentation übernehmen. MHC: Haupthistokompatibilitätskomplex.
Abb. 1 MHC-Moleküle. MHC-Moleküle sind auf kernhaltigen, körpereigenen Zellen (MHC I) und
Antigen-präsentierenden Zellen (MHC II) exprimiert. MHC I-Moleküle bestehen aus einer
α- und einer β-Untereinheit. Die α-Untereinheit enthält drei Domänen, von denen die
Domänen α1 und α2 die Antigenpräsentation übernehmen, und die α3 Domäne die Verankerung in der Zellmembran sichert. Das β2-Mikroglobulin stellt die vierte lösliche Domäne der MHC I-Moleküle dar. MHC II-Moleküle
bestehen aus 2 Untereinheiten, die beide in der Zellmembran verankert sind. Eine Untereinheit
(α- oder β) besteht aus je 2 Domänen, α1 und α2 bzw. β1 und β2, wobei die α1 und die β1 Domäne die Aufgabe der Antigenpräsentation übernehmen. MHC: Haupthistokompatibilitätskomplex.
2.2. Adaptive Immunantwort
Die adaptive Immunantwort ergänzt die angeborene Immunantwort und ermöglicht die Ausbildung
einer persistierenden und Antigen-spezifischen Immunreaktion. Das Prinzip basiert
auf der Antigenpräsentation. Antikörper repräsentieren die humoralen Komponenten der
adaptiven Immunantwort. Sie wirken durch Opsonisierung, Induktion einer Antikörperabhängigen
Zytotoxizität, Neutralisation, Aktivierung des Komplementsystems und Agglutination
der Pathogene. Antikörper werden von B-Zellen produziert und sezerniert. Zusätzlich
zu B-Zellen sind die T-Zellen verantwortlich für die adaptive Immunantwort. Zytotoxische
T-Zellen (CD8+) benötigen dabei zur Aktivierung mehrere Signale: Zum einen muss der
spezifische T-Zell-Rezeptor (TZR) mit einem passenden Antigen in Verbindung kommen
([Abb. 2]). Zusätzlich müssen ko-stimulatorische Rezeptoren der T-Zellen aktiviert werden,
um eine Proliferation sowie eine zytotoxische Zellaktivität zu erreichen. Aufgrund
ihrer Fähigkeit, hochspezifisch zytotoxisch zu wirken, sind CD8+ T-Zellen im besonderen
Fokus neuer onkologischer Therapieansätze: Zum einen im Bereich der adaptiven T-Zell
Therapie, bei der Tumorantigen-spezifische T-Zellen vermehrt und dem Patienten (wieder)
verabreicht werden [7], zum anderen im Bereich der Checkpoint-Inhibitoren, bei der spezifische inhibitorische
Rezeptoren der T-Zellen blockiert werden, um so eine T-Zell-Anergie zu verhindern
[8]. T-Helfer-Zellen (CD4+) sind insbesondere an regulatorischen Prozessen der Immunabwehr
beteiligt. Sie haben dabei meist keine eigene zytotoxische Wirkung, sondern vermitteln
diese über Partnerzellen, bspw. zytotoxische T-Zellen oder NK-Zellen. Die Antigenpräsentation
erfolgt dabei über MHC II-Moleküle. Im Gegensatz zu CD8+ T-Zellen benötigen CD4+ T-Zellen
deutlich häufigeren Antigenkontakt, um stimuliert zu werden. Auch bei CD4+ Zellen
kennt man verschiedene Phänotypen: Der TH1-Phänotyp sezerniert IL2 sowie IFNγ und stimuliert die antitumorale Immunantwort,
wohingegen TH2-Zellen IL4 und IL10 produzieren, die wiederum inhibitorische Wirkung auf das Immunsystem
im Tumormilieu entfalten. Die Berücksichtigung dieser grundlegenden Phänotypen und
deren Einflüsse auf das Tumormilieu ist für die Immuntherapie von entscheidender Bedeutung.
 Abb. 2 Antigenpräsentation und Antigen-Erkennung. CD8+T-Zellen erkennen Antigene, die auf
MHC I-Molekülen präsentiert werden. CD4+T-Zellen erkennen Antigene von Antigenpräsentierenden
Zellen auf MHC II-Molekülen. MHC I-Moleküle werden mit Peptidfragmenten aus dem Zytosol
der Zelle beladen, die permanent aus Abbauprodukten der Proteasomen im Zellinneren
entstehen. Dies dient zur Kontrolle, ob die Zelle körpereigene oder körperfremde Proteine
produziert. MHC-II Moleküle werden ausschließlich auf Antigenpräsentierenden Zellen
exprimiert und präsentieren fremde, extrazelluläre Peptidfragmente, die über Phagozytose
oder Endozytose in die Zelle aufgenommen wurden, um Rezeptoren Antigen-spezifischer
Zellen zu aktivieren. Zusätzlich zur Aktivierung über den TZR/MHC-Komplex (Signal
1) benötigen die T-Zellen ein ko-stimulatorisches Signal (CD80, Signal 2) um aktiviert
zu werden. MHC: Haupthistokompatibilitätskomplex; TZR: T-Zell-Rezeptor.
Abb. 2 Antigenpräsentation und Antigen-Erkennung. CD8+T-Zellen erkennen Antigene, die auf
MHC I-Molekülen präsentiert werden. CD4+T-Zellen erkennen Antigene von Antigenpräsentierenden
Zellen auf MHC II-Molekülen. MHC I-Moleküle werden mit Peptidfragmenten aus dem Zytosol
der Zelle beladen, die permanent aus Abbauprodukten der Proteasomen im Zellinneren
entstehen. Dies dient zur Kontrolle, ob die Zelle körpereigene oder körperfremde Proteine
produziert. MHC-II Moleküle werden ausschließlich auf Antigenpräsentierenden Zellen
exprimiert und präsentieren fremde, extrazelluläre Peptidfragmente, die über Phagozytose
oder Endozytose in die Zelle aufgenommen wurden, um Rezeptoren Antigen-spezifischer
Zellen zu aktivieren. Zusätzlich zur Aktivierung über den TZR/MHC-Komplex (Signal
1) benötigen die T-Zellen ein ko-stimulatorisches Signal (CD80, Signal 2) um aktiviert
zu werden. MHC: Haupthistokompatibilitätskomplex; TZR: T-Zell-Rezeptor.
2.3. Tumorentstehung und Tumorevasion
In der Mitte des 20. Jahrhunderts stellten Burnet und Thomas die Hypothese der „Immunüberwachung“
(engl. Immunosurveillance) auf [9]. Sie besagt, dass im Laufe des Lebens die Häufigkeit nicht-vererbbarer, genetischer
Veränderungen in den Zellen zunimmt. Da dies eine Malignitätsentwicklung begünstige,
müsse ein Überwachungsmechanismus mit immunologischem Hintergrund zur Elimination
oder Inaktivierung dieser mutierten Zellen existieren [10]. Die Hypothese der Immunüberwachung ist mittlerweile zum Modell des „Cancer Immunoediting“
erweitert worden. Dabei werden zeitliche Aspekte in der Tumorentstehung berücksichtigt
und verschiedene Phasen der Balance zwischen Abwehr und Progress beschrieben ([Abb. 3]): Die Eliminierungsphase (engl. „Elimination“), die Gleichgewichtsphase (engl. „Equilibrium“)
und die Entziehungsphase (engl. „Escape“) [11]. In der Eliminierungsphase können das adaptive und das angeborene Immunsystem Tumorzellen
erkennen und zerstören. Die Gleichgewichtsphase repräsentiert die Übergangsphase zwischen
Eliminierungs- und Entziehungsphase, in der das Immunsystem die Tumorzellen unter
Kontrolle hält. Dabei herrscht ein Gleichgewicht zwischen den Interleukinen IL12 (immunstimulierend)
und IL23 (immunsupprimierend) [12]. In der Entziehungsphase verliert das Immunsystem die Kontrolle über den Tumor,
und es kommt zum Tumorprogress: Tumorzellen erreichen dies durch Mechanismen, die
die Erkennung durch das Immunsystem reduzieren [13]
[14]
[15]
[16]
[17]
[18], die zu einer erhöhten Resistenz der Tumorzellen führen [19]
[20] und die eine Inaktivierung von Antitumor-Effektorzellen bewirken [21]. Zusätzlich wird das Tumormilieu zugunsten immunsuppressiver Signalwege beeinflusst.
 Abb. 3 Immunoediting. a Eliminierungsphase: In der Eliminierungsphase erkennen und zerstören das adaptive
und das native Immunsystem Tumorzellen. b Gleichgewichtsphase: In der Gleichgewichtsphase hält das Immunsystem die Tumorzellen
unter Kontrolle, eine vollständige Elimination findet nicht mehr statt. c Entziehungsphase: In der Entziehungsphase verliert das Immunsystem die Kontrolle
über die Tumorzellen mit darauffolgendem Tumorprogress. Der Übergang von Eliminierungsphase
über die Gleichgewichtsphase zur Entziehungsphase wird als Immunoediting bezeichnet.
DC: Dendritische Zelle; T: T-Zellen; G: Granulozyten; M: Makrophagen.
Abb. 3 Immunoediting. a Eliminierungsphase: In der Eliminierungsphase erkennen und zerstören das adaptive
und das native Immunsystem Tumorzellen. b Gleichgewichtsphase: In der Gleichgewichtsphase hält das Immunsystem die Tumorzellen
unter Kontrolle, eine vollständige Elimination findet nicht mehr statt. c Entziehungsphase: In der Entziehungsphase verliert das Immunsystem die Kontrolle
über die Tumorzellen mit darauffolgendem Tumorprogress. Der Übergang von Eliminierungsphase
über die Gleichgewichtsphase zur Entziehungsphase wird als Immunoediting bezeichnet.
DC: Dendritische Zelle; T: T-Zellen; G: Granulozyten; M: Makrophagen.
2.4. Tumormilieu
Fortschritte in der Tumortherapie der letzten Jahrzehnte sind insbesondere auf ein
profunderes Verständnis der wechselseitigen Einflüsse im Tumormilieu zurückzuführen.
Unbestreitbar ist, dass Tumorzellen von der Umgebung beeinflusst werden und vice versa
das Tumormilieu beeinflussen. Immer deutlicher wird, dass die Interaktionen zwischen
den beteiligten Zellen hochkomplex sind, und eine Unterscheidung zwischen Ursache
und Wirkung im Tumormilieu eine wissenschaftliche Herausforderung darstellt. Daher
ist eine genaue Betrachtung der beteiligten (zellulären) Strukturen und Signalwege
dringend geboten. Im Tumormilieu treffen Tumorzellen mit den Zellen und Strukturen
aus dem umgebenden Gewebe aufeinander. Diese beinhalten zusätzlich zu den Zellen des
Immunsystems Komponenten der extrazellulären Matrix, Gefäßstrukturen, Stromazellen
und Fibroblasten. Daher interagieren die Tumorzellen mit vielen, teils wechselnden
Partnern. Aufgrund der heterogenen Zusammensetzung ist das Tumormilieu von Patient
zu Patient unterschiedlich. Zusätzlich zur interindividuellen Varianz besteht eine
intraindividuelle Dynamik. Dieser Faktor wird in der Analyse von Tumor-/ Patientenproben
häufig noch vernachlässigt.
2.4.1. Extrazelluläre Matrix
Die Extrazelluläre Matrix (EZM) beschreibt den Raum außerhalb der zellulären Plasmamembranen
und wird durch die interstitiellen Makromoleküle gebildet. Zu diesen Makromolekülen
gehören Glykoproteine und Polysaccharide. Zusätzlich enthält die EZM im Wesentlichen
Wasser, Elektrolyte und Nährstoffe. Sie wird durch die umliegenden Zellen beeinflusst.
Dadurch ist die Zusammensetzung der EZM nicht unveränderlich, sondern durch Stoffaustausch
und Produktion sowie Abbau der Makromoleküle einem wechselnden Prozess unterworfen.
Neben wichtigen Einflüssen auf die Gewebeeigenschaften der Formgebung, Elastizität
und dem Wassergehalt hat die EZM auch eine wichtige regulatorische Funktion in der
Beeinflussung von Immunreaktionen und der Wundheilung sowie bei der Signaltransduktion
und Bindung von Signalrezeptoren. Dadurch kann eine Beeinflussung der intrazellulären
Genexpression erfolgen, die Auswirkungen auf die Adhäsion, Migration und Proliferation
der umliegenden Zellen zur Folge haben kann. Dies macht deutlich, welchen wichtigen
Einfluss die EZM auf das umliegende Gewebe nimmt. Auch in Tumoren beeinflusst die
EZM das Tumormilieu und die Tumorentwicklung [22]. Dabei spielt die Deregulierung der EZM eine wichtige Rolle. Durch erhöhte Kollagenimplementierung
in die EZM kann eine Integrin-vermittelte Zellproliferation induziert werden [23]. Durch anti-apoptotische Effekte [24] und Unterstützung onkogener Zelltransformationen [25] kann die EZM somit die Grundlage für einen Tumorprogress bilden. Zusätzlich kann
ein Tumorprogress durch inhibierende Einflüsse der EZM auf das Immunsystem verursacht
werden [26]. So können T-Zellen in ihrer Proliferation gehindert werden, bspw. durch Bindung
von LAIR (Leukozyten assoziierten Immunglobulin-ähnlichen Rezeptor, engl. leukocyte-associated
Ig-like receptor) mittels Kollagen I [27] oder durch Kompromittierung Antigen-präsentierender Zellen [28].
2.4.2. Gefäßversorgung
Wie alle stoffwechselaktiven Gewebe sind insbesondere Tumorzellen mit ihrer hohen
Zellteilungsaktivität und dem stark aktivierten Stoffwechsel davon abhängig, mit Nährstoffen
versorgt werden zu können. Auch der Sauerstoffverbrauch und der Abtransport von Stoffwechselmetaboliten
sind wichtige Gründe für die Abhängigkeit der Tumoren von einer adäquaten Gefäßversorgung.
Daher ist die Angiogenese im Tumormilieu wichtig für das Tumorwachstum, das Invasions-
und Metastasierungsverhalten [29]
[30]. Die Angiogenese beschreibt den Prozess der Gefäßneubildung, die aus dem bereits
bestehenden Gefäßbett heraus entsteht. Anreize für die Angiogenese werden unter anderem
durch Hypoxie, einem niedrigen pH-Wert, Hypoglykämie, Stress und Entzündung hervorgerufen.
Dabei werden Endothelzellen durch Wachstumsfaktoren aktiviert, die dann migrieren
und proliferieren. Zu diesen Wachstumsfaktoren gehören die vaskulären endothelialen
Wachstumsfaktoren (VEGF, engl. Vascular Endothelial Growth Factor), die Fibroblasten-Wachstumsfaktoren
(FGF, engl. Fibroblast Growth Factor), die Plättchen-Wachstumsfaktoren (PDGF, engl.
Platelet Derived Growth Factor), die Hepatozyten-Wachstumsfaktoren (HGF, engl. Hepatocyte
Growth Factor) und Angiopoetin 1 und 2 [30]. Die Rezeptoren dieser Wachstumsfaktoren, vornehmlich Tyrosinkinaserezeptoren, können
dabei nicht nur durch ihre Liganden, sondern auch durch Hormone, Neurotransmitter
und Lymphokine stimuliert werden. Gerade Letztere nehmen in der Angiogenese einen
wichtigen Platz ein, denn die beteiligten Botenstoffe spielen auch bei Inflammation
und Immunzellmigration eine wichtige Rolle [29]. So können bspw. Mastzellen und Makrophagen durch Tumorzellen rekrutiert und zur
Sekretion angiogenetisch-wirksamer Zytokine stimuliert werden.
2.4.3. Fibroblasten
Fibroblasten sind mesenchymalen Ursprungs und als Zellen des Bindegewebes am Aufbau
der EZM beteiligt. Sie sezernieren hierfür v. a. Kollagen, Fibronektin und Wachstumsfaktoren.
Gleichzeitig sind sie allerdings auch an Umstrukturierungen der EZM beteiligt und
können dafür Matrixmetalloproteasen (MMP) herstellen, die Peptidbindungen von Strukturen
der EZM lösen und somit eine wichtige Rolle in der Angiogenese, der Wundheilung und
dem Tumorwachstum übernehmen. Als Zellen mesenchymalen Ursprungs sind sie somit auch
Bestandteil des Tumormilieus solider Tumoren. Mesenchymale Stromazellen können im
Tumormilieu zu sogenannten Krebs-assoziierten Fibroblasten (CAF, engl. Cancer Associated
Fibroblasts) differenzieren. Diese weisen eine deutlich erhöhte Aktivität im Vergleich
zu anderen Fibroblasten auf [31]. Dabei sezernieren sie TGFβ und andere Wachstumsfaktoren, die fördernd auf das Tumorwachstum
wirken [32]. Auch die metabolischen Prozesse in Tumorzellen können durch die CAFs unterstützt
werden [33]. Zusätzlich werden die durch CAF sezernierten MMPs mit erhöhter Invasivität und
Metastasierung in Verbindung gebracht [32]
[34]. Auf das Immunsystem wirken die CAF durch chronische Zytokinsekretion, die eine
Ursache der chronischen Inflammationsreaktion im Tumormilieu darstellt. Die mobilisierten
Immunzellen wie bspw. Makrophagen werden durch die chronische Stimulation der CAF
und weitere immunmodulierende Einflüsse des Tumormilieus im Rahmen der chronischen
Inflammationsreaktion zum tumorfördernden Phänotyp (M2) konvertiert [35]
[36].
2.4.4. Suppressive Immunzellen
Zusätzlich zu den eingangs beschriebenen zellulären Anteilen des Immunsystems gibt
es weitere Immunzellen, die insbesondere im Tumormilieu vorhanden sind: Regulatorische
T-Zellen (Treg), Tumor-assoziierte Makrophagen (TAM) und Myeloide Suppressorzellen
(MDSC, engl. Myeloid-Derived Suppressor Cell).
2.4.4.1. Treg Treg stellen hauptsächlich eine Subpopulation von CD4+ T-Zellen dar, die als sogenannte
Suppressorzellen für die Erhaltung der Selbsttoleranz verantwortlich sind. Dies wird
durch Inhibition aktivierter T-Zellen erreicht. Treg zeichnen sich durch die Expression
von CD4 und CD25 (Untereinheit des IL2 Rezeptors) und des Transkriptionsfaktors FOXP3
(forkhead box protein 3) aus. Bisher sind über vier unterschiedliche CD4+ Subpopulationen
von regulatorischen T-Zellen beschrieben worden. Die unterschiedlichen Subpopulationen
können gegebenenfalls für die z. T. widersprüchlich erscheinenden Forschungsergebnisse
verantwortlich sein. Trotz dessen besteht der weit verbreitete Konsens, dass Treg
im Tumormilieu immunsuppressive Eigenschaften aufweisen [37]. Sie vermitteln diese Wirkung nicht nur durch Sekretion spezifischer Interleukine
und anderer Zytokine, sondern benötigen für einen Teil ihrer Funktionen auch direkten
Zell-zu-Zellkontakt [38]. Die beteiligten Zytokine sind hauptsächlich IL4, IL10, IL35 und TGFβ. Eine Übersicht
der Zytokineinflüsse auf CD4+ T-Zellen bietet [Abb. 4]. Im direkten Zell-zu-Zell Kontakt vermitteln Treg immunsuppressive Eigenschaften
über die Expression sogenannter Checkpoint-Moleküle, insbesondere CTLA4 und LAG3 (engl.
Lymphocyte-Activation Gene 3), sowie Oberflächen-gebundene Enzyme wie CD39. Die Checkpoint-Moleküle
wirken dabei über eine kompetitive Bindung im Zell-zu-Zell Kontakt mit ko-stimulierenden
Molekülen von Antigen präsentierenden Zellen und verhindern so eine effektive Antigenpräsentation.
Zusätzlich wird die Reifung der Antigen präsentierenden Zellen, insbesondere von Dendritischen
Zellen verhindert. Die enzymatische Funktion von CD39, einer Ektonukleotidase, spielt
bei der Verarbeitung von ATP zu AMP eine wichtige Rolle. Die pro-inflammatorischen
Eigenschaften von extrazellulärem ATP werden dadurch inhibiert. Neben den Dendritischen
Zellen haben Treg insbesondere eine inhibitorische Wirkung auf aktivierte CD4+ und
CD8+ T-Zellen. Sie tragen wesentlich zu Entwicklung einer T-Zell-Anergie bei [39]. Im anergen Zustand können die T-Zellen dabei nicht mehr stimuliert werden und reagieren
auch nicht auf eine erneute Antigenpräsentation. Auch die Proliferation der T-Zellen
wird über eine inhibitorische Wirkung auf den Zellzyklus verhindert [39]. Zusätzlich stehen die Treg in Kompetition mit den anderen T-Zellen um IL2, welches
für die Proliferation und Aktivierung sowohl der Treg als auch der anderen T-Zellen
im Tumormilieu benötigt wird. Auch auf die NK-Zellen konnte eine inhibitorische Wirkung
der Treg nachgewiesen werden. Die Sekretion von TGFβ bewirkt eine verminderte Expression
von NKG2D, einem aktivierenden NK-Zellrezeptor [40]. Die Möglichkeiten der Treg, auf die Immunantwort Einfluss zu nehmen, sind daher
sehr vielfältig und im Tumormilieu besonders deutlich ausgeprägt [41].
 Abb. 4 Der Einfluss des Zytokinmilieus auf die Differenzierung naiver CD4+ T-Zellen. CD4+
T-Zellen werden durch unterschiedliche Zytokine zur Differenzierung in TH1 und TH2 sowie TH17 und Treg Zellen beeinflusst. Dabei induziert insbesondere TGFβ die Entwicklung
von Treg. Treg selbst beeinflussen das Tumormilieu mittels TGFβ und IL10. Zusätzlich
unterscheidet man bei der Entwicklung von T-Zellen die Differenzierung in TH1, TH2 und TH17 Zellen. IL12 stimuliert eine Differenzierung in TH1-Zellen, die mit einer antitumoralen Antwort im Tumormilieu assoziiert sind. IL4
ist für die Induktion von TH2-Zellen wichtig, die eine protumorale Rolle im Tumormilieu mittels IL4, IL10 und
IL13 Sekretion übernehmen. Die Rolle der TH17-Zellen im Tumormilieu ist noch umstritten, sie werden durch IL6 und IL23 und TGFβ
stimuliert. Grün: vorwiegend antitumorale Wirkung; Rot: vorwiegend protumorale Wirkung;
IL: Interleukin; TGF: Transformierender Wachstumsfaktor; IFN: Interferon; TNF: Tumornekrosefaktor;
T-bet: T-box Transkriptionsfaktor; GATA3: GATA Transkriptionsfaktor; FoxP3: F box
protein 3-Transkriptionsfaktor.
Abb. 4 Der Einfluss des Zytokinmilieus auf die Differenzierung naiver CD4+ T-Zellen. CD4+
T-Zellen werden durch unterschiedliche Zytokine zur Differenzierung in TH1 und TH2 sowie TH17 und Treg Zellen beeinflusst. Dabei induziert insbesondere TGFβ die Entwicklung
von Treg. Treg selbst beeinflussen das Tumormilieu mittels TGFβ und IL10. Zusätzlich
unterscheidet man bei der Entwicklung von T-Zellen die Differenzierung in TH1, TH2 und TH17 Zellen. IL12 stimuliert eine Differenzierung in TH1-Zellen, die mit einer antitumoralen Antwort im Tumormilieu assoziiert sind. IL4
ist für die Induktion von TH2-Zellen wichtig, die eine protumorale Rolle im Tumormilieu mittels IL4, IL10 und
IL13 Sekretion übernehmen. Die Rolle der TH17-Zellen im Tumormilieu ist noch umstritten, sie werden durch IL6 und IL23 und TGFβ
stimuliert. Grün: vorwiegend antitumorale Wirkung; Rot: vorwiegend protumorale Wirkung;
IL: Interleukin; TGF: Transformierender Wachstumsfaktor; IFN: Interferon; TNF: Tumornekrosefaktor;
T-bet: T-box Transkriptionsfaktor; GATA3: GATA Transkriptionsfaktor; FoxP3: F box
protein 3-Transkriptionsfaktor.
2.4.4.2 MDSC MDSC ist der Überbegriff für eine Subgruppe von Suppressorzellen, die myeloiden Ursprungs
sind. Sie werden durch die Expression spezifischer Oberflächenmoleküle definiert (CD11b+,
CD33+ und CD34+) und konnten in den meisten soliden Tumoren nachgewiesen werden. Sie
zeichnen sich insbesondere durch eine protumorale Differenzierung aus. Im Gegensatz
zu reifen Dendritischen Zellen supprimieren MDSC die Immunantwort im Tumormilieu ([Abb. 5]). MDSC werden durch pro-inflammatorische Signale stimuliert und sind daher besonders
relevant in der Steuerung der Immunantwort im Tumormilieu [42]. Ähnlich wie Treg üben MDSC ihre immunsupprimierende Wirkung in vielen Bereichen
der adaptiven und der angeborenen Immunantwort aus. Dabei bedienen sie sich unterschiedlicher
Mechanismen: T-Zellen werden in der Antigenerkennung durch Nitrierung des T-Zell-Rezeptors
gestört. Außerdem verhindern sie die T-Zell-Aktivierung über den Verbrauch von Cystein,
einer für T-Zellen essentiellen Aminosäure [43]. Auch die Proliferation von T-Zellen wird über eine Hemmung der IL2-Produktion beeinflusst.
Die Produktion von Arginase und Sauerstoffradikalen behindert die Antigenerkennung
und die T-Zell-Aktivierung [44]. Zusätzlich beeinflusst die vermehrte Differenzierung zu MDSC aus Vorläuferzellen
myeloiden Ursprungs die adaptive Immunantwort über eine Reduktion antigenpräsentierender
Zellen zugunsten der MDSC. Darüber hinaus unterstützen MDSC die Bildung von Tregs
über die vermehrte Produktion von IL10, TGFβ und Arginase [42]. Auch Teile der angeborenen Immunantwort werden durch die MDSC gehemmt, insbesondere
NK-Zellen und M1-Makrophagen, die über eine vermehrte Produktion von IL10 und eine
Verminderung der IL12-Sekretion in ihrer Funktion gestört werden [45]. Die Entwicklung von MDSC wird durch verschiedene Zytokine beeinflusst, darunter
VEGF, Granulozyten- und Monozytenkolonie-stimulierender Faktor (GM-CSF, engl. Granulocyte
Monocyte Colony-Stimulating Factor), IL6, IL1β, PGE2 (ProstaGlandin E2) und Komplement
C5a.
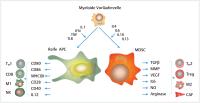 Abb. 5 Myeloide Suppressorzellen und Antigenpräsentierende Zellen im Tumormilieu. MDSC werden
durch pro-inflammatorische Signale stimuliert und supprimieren die Immunantwort im
Tumormilieu. Sie induzieren die Bildung von Treg und TH2-Zellen und fördern die Differenzierung zu M2-Phänotypen. Reife Antigenpräsentierende
Zellen können die antitumorale Immunantwort stimulieren, indem sie durch Antigenpräsentierende
und Ko-Stimulation CD8+ T-Zellen, CD4+ TH1-Zellen und NK-Zellen unterstützen. Grün: vorwiegend antitumorale Wirkung; Rot: vorwiegend
protumorale Wirkung; IL: Interleukin; TGF: Transformierender Wachstumsfaktor; IFN:
Interferon; TNF: Tumornekrosefaktor; M1: M1 Phänotyp; NK: Natürliche Killerzelle;
TLR: Toll-like-Rezeptor-Stimulation; MMP: Matrixmetalloprotease; VEGF: vaskulärer
endothelialer Wachstumsfaktor; NO: Stickoxid.
Abb. 5 Myeloide Suppressorzellen und Antigenpräsentierende Zellen im Tumormilieu. MDSC werden
durch pro-inflammatorische Signale stimuliert und supprimieren die Immunantwort im
Tumormilieu. Sie induzieren die Bildung von Treg und TH2-Zellen und fördern die Differenzierung zu M2-Phänotypen. Reife Antigenpräsentierende
Zellen können die antitumorale Immunantwort stimulieren, indem sie durch Antigenpräsentierende
und Ko-Stimulation CD8+ T-Zellen, CD4+ TH1-Zellen und NK-Zellen unterstützen. Grün: vorwiegend antitumorale Wirkung; Rot: vorwiegend
protumorale Wirkung; IL: Interleukin; TGF: Transformierender Wachstumsfaktor; IFN:
Interferon; TNF: Tumornekrosefaktor; M1: M1 Phänotyp; NK: Natürliche Killerzelle;
TLR: Toll-like-Rezeptor-Stimulation; MMP: Matrixmetalloprotease; VEGF: vaskulärer
endothelialer Wachstumsfaktor; NO: Stickoxid.
2.4.4.3. TAM Aufgrund ihrer Vielseitigkeit, Mobilität und der Zugehörigkeit zum angeborenen Immunsystem
sind Makrophagen in vielen Pfaden der Immunabwehr − einschließlich Wundheilung und
Entzündungsprozessen − aktiv. Dies schließt auch das Tumormilieu mit ein, in dem Makrophagen
an der Angiogenese, der Leukozyteninfiltration, der Veränderung der EZM und der Immunsuppression
beteiligt sind [46]. In Zusammenhang mit dem Tumormilieu ist dabei in den letzten Jahren eine heterogene
Gruppe von Makrophagen beschrieben worden, die als Tumor-assoziierte Makrophagen bezeichnet
wird. Dabei ist zu beachten, dass einige Autoren in Zusammenhang mit Tumoren vornehmlich
den M2-Phänotyp beschreiben, andere aufgrund der hohen Plastizität der Makrophagen
auch bei TAM zwischen dem M1-und dem M2-Phänotyp unterscheiden ([Abb. 6]). In der überwiegenden Mehrzahl der Tumoren (bis auf das Kolonkarzinom) korrelieren
TAM mit einer schlechten Prognose [47]. Dabei können sie bis zu einem Drittel der zellulären Bestandteile des Tumormilieus
ausmachen [48]. TAM werden über Chemokine (z. B. Chemokinligand 2, CCL2 und CCL5), die meist von
Tumor- oder Stromazellen sezerniert werden, in das Tumormilieu rekrutiert. Außerdem
sind VEGF, PDGF, M-CSF und TGFβ an der Makrophagenrekrutierung beteiligt [49]. M1-Makrophagen werden durch IFNγ, LPS, GM-CSF und TNF polarisiert, produzieren
viel IL1, IL6, IL12, IL23, TNF und niedrige Level an IL10 und bewirken eine TH1-Antwort, Gewebezerstörung und Immunstimulation. M2-Makrophagen werden v. a. durch
IL4, IL10 und IL13 polarisiert, produzieren viel IL10, TGFβ und wenig IL1, IL6, IL12
und TNF und bewirken eine TH1-Suppression, TH2-Aktivierung, Immunsuppression und fördern die Wundheilung und Geweberegeneration.
Da Makrophagen eine hohe Plastizität aufweisen und stark von den Einflüssen des Tumormilieus
abhängen, sind die TAM daher eher dem M2-Phänotyp zugeordnet [5]. Daher korrelieren sie im Tumor mit der Angiogenese, der Bildung von Metastasen
und dem Tumorprogress [6].
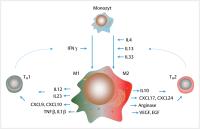 Abb. 6 Monozyten im Tumormilieu. Monozyten differenzieren im Tumormilieu in M1- und M2-Makrophagen
in Abhängigkeit der vorherrschenden Einflüsse. M1-Makrophagen stimulieren über IL12
die TH1-Antwort und sind mit einer antitumoralen Wirkung assoziiert. M2-Makrophagen stimulieren
eine TH2-Antwort und wirken durch die Bildung von IL10, VEGF und Arginase protumoral im Tumormilieu.
Grün: vorwiegend antitumorale Wirkung; Rot: vorwiegend protumorale Wirkung; IL: Interleukin;
IFN: Interferon; TNF: Tumornekrosefaktor; M1: M1 Phänotyp; CXCL: Chemokin; VEGF: vaskulärer
endothelialer Wachstumsfaktor; EGF: epidermaler Wachstumsfaktor.
Abb. 6 Monozyten im Tumormilieu. Monozyten differenzieren im Tumormilieu in M1- und M2-Makrophagen
in Abhängigkeit der vorherrschenden Einflüsse. M1-Makrophagen stimulieren über IL12
die TH1-Antwort und sind mit einer antitumoralen Wirkung assoziiert. M2-Makrophagen stimulieren
eine TH2-Antwort und wirken durch die Bildung von IL10, VEGF und Arginase protumoral im Tumormilieu.
Grün: vorwiegend antitumorale Wirkung; Rot: vorwiegend protumorale Wirkung; IL: Interleukin;
IFN: Interferon; TNF: Tumornekrosefaktor; M1: M1 Phänotyp; CXCL: Chemokin; VEGF: vaskulärer
endothelialer Wachstumsfaktor; EGF: epidermaler Wachstumsfaktor.
2.4.5. Tumorstammzellen
Als Tumorstammzellen (CSC, engl. Cancer Stem Cells) werden Tumorzellen mit Stammzelleigenschaften
bezeichnet. Zu diesen Eigenschaften gehören die Selbsterneuerung sowie das Potenzial
zur Differenzierung [50]. Die Hypothese der Tumorstammzellen ist zwar umstritten [51], es verdichten sich jedoch die Hinweise, dass in unterschiedlichen Tumorentitäten
Tumorzellen mit Stammzelleigenschaften gefunden werden [52]. Tumorstammzellen werden z. T. für die Ausbildung von Therapieresistenzen verantwortlich
gemacht [53]. In Verbindung mit Chemotherapie konnte zuletzt eine Konversion von Glioblastomzellen
in Tumorstammzellen gezeigt werden [54]. Eine weitere wichtige Eigenschaft von Tumorstammzellen ist die niedrige Immunogenität
[55]. Die Fähigkeit, dem Immunsystem auszuweichen, basiert auf unterschiedlichen Eigenschaften.
Zum einen exprimieren CSC inhibitorische Liganden, wie z. B. FasL und inhibitorische
NK Liganden, zum anderen anti-apototische Moleküle wie Bcl2 und Survivin. Außerdem
sezernieren sie die klassischen immunsupprimierenden Zytokine wie TGFβ, IL4, IL6,
IL10 und PGE2. Zusätzlich inhibieren CSC die T-Zell-Proliferation STAT3-vermittelt
[56].
3. Immuntherapien
Ziel der unterschiedlichen Immuntherapien ist es, die Aktivität des Immunsystems möglichst
gezielt gegen den Tumor zu richten, protumorale Effekte aufzuheben und eine Tumorelimination
zu ermöglichen ([Abb. 7]). Auf die Grundlagen der pro- und antitumoralen Prozesse und die beteiligten zellulären
und nichtzellulären Strukturen wurde in den vorherigen Kapiteln eingegangen. Im Folgenden
sollen daher die einzelnen therapeutischen Ansätze hinsichtlich ihrer Wirkungsweisen
genauer betrachtet werden.
 Abb. 7 Übersicht über pro- und antitumorale Einflüsse im Tumormilieu. Durch den Einfluss
des Tumors wird ein protumorales Tumormilieu geschaffen und damit werden Zelltypen
stimuliert, die ihrerseits die Tumorentwicklung begünstigen. Die verschiedenen Ansätze
der Immuntherapien unterstützen die Bildung eines antitumoralen Milieus mit dem Ziel
der Tumorelimination. Grün: vorwiegend antitumorale Wirkung; Rot: vorwiegend protumorale
Wirkung; IL: Interleukin; IFN: Interferon; TNF: Tumornekrosefaktor; M1: M1-Phänotyp;
M2: M2-Phänotyp; MDSC: Myeloide Suppressorzelle; Treg: regulatorische T-Zelle; NK:
Natürliche Killerzelle; DC: Dendritische Zelle; CD: Cluster of Differentiation; HLA-DR:
Humane Leukozytenantigen-DR; CAF: Krebs-assoziierter Fibroblast; TGF: Transformierender
Wachstumsfaktor.
Abb. 7 Übersicht über pro- und antitumorale Einflüsse im Tumormilieu. Durch den Einfluss
des Tumors wird ein protumorales Tumormilieu geschaffen und damit werden Zelltypen
stimuliert, die ihrerseits die Tumorentwicklung begünstigen. Die verschiedenen Ansätze
der Immuntherapien unterstützen die Bildung eines antitumoralen Milieus mit dem Ziel
der Tumorelimination. Grün: vorwiegend antitumorale Wirkung; Rot: vorwiegend protumorale
Wirkung; IL: Interleukin; IFN: Interferon; TNF: Tumornekrosefaktor; M1: M1-Phänotyp;
M2: M2-Phänotyp; MDSC: Myeloide Suppressorzelle; Treg: regulatorische T-Zelle; NK:
Natürliche Killerzelle; DC: Dendritische Zelle; CD: Cluster of Differentiation; HLA-DR:
Humane Leukozytenantigen-DR; CAF: Krebs-assoziierter Fibroblast; TGF: Transformierender
Wachstumsfaktor.
3.1. Zytokintherapien
Zytokine sind Proteine, welche die Aktivität, die Migration und die Differenzierung
von Zellen regulieren. Im Tumormilieu ist die Zytokinsekretion ein wichtiger Kommunikationsweg
zwischen Tumor- und Immunzellen. Die Bedeutung des Zytokinmilieus wird ersichtlich,
wenn man sich vor Augen führt, dass bspw. die Differenzierung – und damit die Funktion
– von CD4+ T-Zellen ganz wesentlich von Zytokinen gesteuert wird. Sie entscheiden
darüber, ob eine TH1-Zelle mit antitumoraler Aktivität entsteht oder eine Treg Zelle mit immunsupprimierender
Funktion [57]. Zur Gruppe der Zytokine gehören die Interleukine, die Chemokine, die Tumornekrosefaktoren
(TNF), die Interferone und die koloniestimulierenden Faktoren (CSF, engl. Colony Stimulating
Factor).
Interleukine sind Peptidhormone, die zu den Zytokinen gehören und Zellwachstum und
-differenzierung beeinflussen und von allen Zellen des Immunsystems gebildet werden.
Sie können sowohl stimulierend, als auch inhibierend auf Wachstum, Teilung und insbesondere
Differenzierung anderer Immunzellen wirken. Aufgrund ihrer meist pleiotropen Wirkung
beeinflussen Interleukine mehrere phänotypische Merkmale. Mittlerweile sind über 40
Interleukine mit unterschiedlichen physiologischen Funktionen bekannt, die ihrer Entdeckung
entsprechend chronologisch beziffert werden. Interleukine wirken im Tumormilieu hauptsächlich
parakrin.
Aufgrund ihrer Funktion bieten sich Interleukine als therapeutisches Mittel an, um
auf die Kommunikation und die Steuerung des Immunsystems Einfluss nehmen zu können.
In den letzten Jahrzehnten sind daher verschiedene immuntherapeutische Ansätze auf
der Basis von Interleukintherapien entwickelt worden [58]. Dazu gehören die Interleukine IL2, IL7, IL12, IL18 und IL21 [57]. Eines der wichtigsten bisher therapeutisch genutzten Interleukine ist IL2. Wie
bereits erwähnt nimmt es eine zentrale Rolle bei der Aktivierung und Proliferation
von T-Zellen ein. Es ist das erste Interleukin, welches im Menschen als Krebsmedikament
angewendet wurde und klinische Erfolge erzielen konnte. Historisch gesehen wurde IL2
– der T-Zell Wachstumsfaktor – 1976 entdeckt [59] und 1994 als Medikament zur Behandlung von metastasierten Nierenzellkarzinomen sowie
1998 in den USA auch für das metastasierte maligne Melanom zugelassen. Die systemische
Anwendung des IL2 gerade bei metastasierten Tumoren birgt aber den Nachteil beträchtlicher
Nebenwirkungen. Unter Umständen kann IL2 zu einem sogenannten Vascular Leak Syndrom
führen, bei dem die Durchlässigkeit der Gefäßwände aufgrund einer endothelzellvermittelten
Hyperpermeabilität stark erhöht ist. Dadurch kommt es zu Paravasaten, die behandlungslimitierende
Folgen haben können [60]. Für einen kleinen Teil (8%, 33 von 409) der behandelten Patienten konnte allerdings
eine komplette, langanhaltende (mediane Beobachtungszeit > 7 Jahre) Remission von
metastasierten Nierenkarzinomen (9,3%) und Melanomen (6,6%) erreicht werden [61]. Weitere Interleukine sind aktuell Gegenstand klinischer Studien [62].
Chemokine wirken auf andere Zellen chemotaktisch und werden im Falle einer Entzündung
oder eines anderen akuten Ereignisses vermehrt freigesetzt. Chemokine werden unter
anderem mit einer erhöhten Angiogenese in Verbindung gebracht [63]. Interessanterweise ist die Chemokinrezeptorexpression in Tumor-infiltrierenden
Lymphozyten herunterreguliert, was mit einer erhöhten Internalisierung der Rezeptoren
aufgrund der starken Konzentration im Tumormilieu begründet wird [64]. In klinischen Studien wurden auch monoklonale Antikörper gegen Chemokinrezeptoren
bei Patienten mit T-Zell Malignomen [65] untersucht. Zusätzlich behandelt eine aktuelle Studie die Chemokinmodulation in
Patienten mit Kolonkarzinomen (NCT01545141).
Tumornekrosefaktoren (TNFα und TNFβ) sind an der Proliferation, Differenzierung, Apoptose,
Nekrose, Angiogenese und Aktivierung des Immunsystems beteiligt. TNF haben zusätzlich
Einfluss auf den Fettstoffwechsel, die Insulinresistenz und die Endothelzellen, wirken
systemisch über Fieber und können Kachexie (TNFα wurde früher als Kachexin bezeichnet)
verursachen. In der Tumortherapie wird TNFα beim Malignen Melanom und bei Sarkomen
des Weichgewebes eingesetzt [66]. Im Tumor selbst induziert es eine Hyperpermeabilität der Gefäße mit darauffolgender
hämorrhagischer Nekrose und Zerstörung der Gefäßstruktur [67]. Dennoch haben sich die initialen Hoffnungen, eine TNFα-basierte Tumortherapie breit
einsetzen zu können, bisher nicht bestätigt. Begründet wird dies z. T. mit der pleiotropen
Charakteristik von TNFα und mit der vom zeitlichen Verlauf abhängenden, unterschiedlichen
Wirkungsweise.
Interferone sind pleiotrope Zytokine, die sowohl von Stromazellen, als auch von Immunzellen
gebildet werden und eine breite immunstimulierende Wirkung über die Aktivierung von
Transkriptionsproteinen (Jak-STAT Signalweg) und eine erhöhte Expression von Komponenten
der Antigenpräsentation (z. B. MHC-Moleküle) besitzen. Physiologisch erfolgt die Interferonbildung
v. a. nach Aktivierung durch virale oder bakterielle Antigene. Im Menschen werden
IFNα, IFNβ (Typ I Interferone) und IFNγ (Typ II Interferon) unterschieden. IFNα und
IFNβ verstärken die MHC I-Expression, aktivieren Dendritische Zellen, T-Zellen und
NK-Zellen [68] und inhibieren die Bildung immunsuppressiver Treg und MDSC [69]
[70]. Zusätzlich besitzen sie eine Wirkung auf Tumorzellen und führen zu einer vermehrten
Differenzierung und einer erhöhten Tumorantigenpräsentation [71]. IFNγ aktiviert insbesondere CD8+ T-Zellen und verstärkt die MHC II-Expression sowie
die Bildung des Makrophagen M1-Phänotyps [68]. Andererseits wurde in den letzten Jahren auch eine Interferon-abhängige immunsupprimierende
Wirkung im Tumormilieu beobachtet. [72] Diese basiert auf einer vermehrten Expression eines Checkpoint-Rezeptor-Liganden
(PDL1, engl. Programmed Death receptor Ligand 1) nach Interferonstimulation. Somit
werden auch inhibierende Effekte vermehrt beobachtet [73]. Dabei kann die Dauer der Stimulation eine wichtige Rolle spielen und den Unterschied
zwischen einer akuten Entzündungsreaktion mit tumorhemmenden Eigenschaften und einer
chronischen Entzündungsreaktion mit tumorfördernden Eigenschaften ausmachen. Dies
hat wesentliche Konsequenzen für den klinischen Einsatz. Die immunaktivierenden Eigenschaften
der Interferone werden therapeutisch eingesetzt, um bspw. virale Hepatitiden (IFNα)
zu behandeln. Aber auch in der Tumortherapie wird die Behandlung mit Interferonen
gegen spezifische Tumorentitäten eingesetzt (z. B. spezielle Lymphome, Leukämien,
Kaposi-Sarkome, Malignes Melanom). Zu beachten ist auch hier, dass eine Limitierung
durch teils starke Nebenwirkungen auftreten kann. Daher gibt es Bestrebungen, die
Wirkung zu optimieren und gleichzeitig die Nebenwirkungen zu reduzieren [74]. Beim Malignen Melanom konnte ein grenzwertig signifikanter Einfluss auf das progressionsfreie
Überleben nach erfolgter Resektion eines Stadium III Melanoms im Vergleich zur klinischen
Beobachtung festgestellt werden [75]. Daher ist der Interferoneinsatz in der Tumortherapie unter anderem von der Tumorentität,
dem Nebenwirkungsspektrum und der ggf. damit verbundenen Patientencompliance abhängig
und muss individuell in Absprache mit dem Patienten erfolgen.
Koloniestimulierende Faktoren sind Glykoproteine, die die Proliferation und Differenzierung
von Zellen beeinflussen können, die dem hämatopoetischen System entstammen. Es werden
mehrere CSF unterschieden: G-CSF (Granulozyten), M-CSF (Monozyten), GM-CSF (Granulozyten
und Monozyten), Meg-CSF (Megakaryozyten) und der SCF (Stammzellfaktor). Außerdem werden
einige Interleukine, z. B. IL2 sowie Erythropoetin, zu den CSF gezählt, da sie ebenfalls
Proliferation und Differenzierung von Zellen des hämatopoetischen Systems beeinflussen.
Die Bildung und Sekretion erfolgt im Knochenmark, im Stroma und in Immunzellen (B
und T-Zellen, Makrophagen u.w.). In der Onkologie werden CSF als Adjuvantien eingesetzt,
um nach Suppression eine Restitution der hämatopoetischen Zelllinien zu erreichen.
Allerdings werden CSF auch in der aktiven Tumorbehandlung untersucht. So ist wie zuvor
beschrieben die Anzahl an intratumoralen TAM mit einer schlechten Prognose assoziiert.
Daher gibt es Bestrebungen, den Rezeptor von M-CSF zu blockieren, um das Überleben
der Makrophagen zu verhindern und somit die Anzahl an TAM zu reduzieren. Phase I und
II Studien zeigten bisher eine limitierte bis moderate Wirkung in der Monotherapie
bei moderatem Nebenwirkungsprofil [76]
[77]. Auch GM-CSF wird in der onkologischen Immuntherapie verwendet, da gezeigt werden
konnte, dass eine Differenzierung der Dendritischen Zellen, eine Suppression der MDSC
und eine Ausbildung des M1-Phänotypes induziert wird [78]
[79].
3.2. Toll-like-Rezeptor Stimulation
„Pattern Recognition“ Rezeptoren (PRR) erkennen Moleküle, die mit Pathogenen wie bspw.
Viren oder Bakterien assoziiert sind (PAMP, Pathogen-assoziierten molekularen Muster).
Diese Rezeptoren initiieren eine Abwehrreaktion und gehören zu den epigenetisch ältesten
Komponenten der Immunantwort. Die Toll-like-Rezeptoren (TLR), eine Hauptgruppe der
PRR, sind daher auch innerhalb der verschiedenen Spezies weit verbreitet und wurden
Mitte der 1990er Jahre erstmals bei Drosophila melanogaster entdeckt. Im Menschen sind seitdem 10 verschiedene TLR-Subtypen identifiziert worden.
Die Aktivierung der TLR hat eine intrazelluläre Signalkaskade zur Folge ([Abb. 8]), die im Wesentlichen eine Differenzierung von Zellen myeloiden Ursprungs bewirkt
und deren Reifung und Proliferation stimuliert. Zusätzlich werden in der Folge auch
Zellen der adaptiven Abwehr aktiviert. Die immunologisch aktivierende Wirkung einzelner
TLR-Agonisten wurden in verschiedenen Krebsentitäten untersucht und führte zur Zulassung
einzelner Immuntherapeutika dieser Wirkstoffklasse. Dazu gehören die TLR2/4-Agonisten
Bacillus Calmette-Guérin (BCG, inaktiviertes Mykobakterium Bovis) bei Blasenkarzinomen,
der TLR4-Agonist Picibanil bei Kopf-Hals-Karzinomen [80], oder der TLR7-Agonist Imiquimod bei Basalzellkarzinomen. Resiquimod, ein potenterer
TLR7-und TLR8-Agonist ist aktuell als Nachfolger in der klinischen Entwicklung. Ein
weiterer TLR8-Agonist, der zurzeit in der Klinik erforscht wird, ist Motolimod bei
Kopf-Hals-Karzinomen und anderen soliden Tumoren (NCT01836029). Außerdem werden TLR-Agonisten
mittlerweile aufgrund ihrer Förderung der Reifung und Ausdifferenzierung antigenpräsentierender
Zellen häufig mit Vakzinierungen zur Unterstützung der Antigenpräsentation kombiniert
[81].
 Abb. 8 Toll-like-Rezeptoren. Toll-like-Rezeptoren können sowohl auf der Zelloberfläche als
auch intrazellulär gebunden werden. Ihre Aktivierung hat eine Signalkaskade zur Folge,
die hauptsächlich über MyD88 und NFKB eine Produktion von IFNγ, TNFα und IL12 initiiert. Zusätzlich wird eine Zelldifferenzierung
stimuliert. NFKB: Transkriptionsfaktor; MyD88: Myeloides Differenzierungsprotein 88; TIR: Toll/IL-1 R
Homologie Domäne; TIRAP: Adaptermolekül; IRAK: Interleukin-1 Rezeptor assoziierte
Kinase; TRAF: TNF Rezeptor-assoziierter Faktor; IFN: Interferon; TNF: Tumornekrosefaktor.
Abb. 8 Toll-like-Rezeptoren. Toll-like-Rezeptoren können sowohl auf der Zelloberfläche als
auch intrazellulär gebunden werden. Ihre Aktivierung hat eine Signalkaskade zur Folge,
die hauptsächlich über MyD88 und NFKB eine Produktion von IFNγ, TNFα und IL12 initiiert. Zusätzlich wird eine Zelldifferenzierung
stimuliert. NFKB: Transkriptionsfaktor; MyD88: Myeloides Differenzierungsprotein 88; TIR: Toll/IL-1 R
Homologie Domäne; TIRAP: Adaptermolekül; IRAK: Interleukin-1 Rezeptor assoziierte
Kinase; TRAF: TNF Rezeptor-assoziierter Faktor; IFN: Interferon; TNF: Tumornekrosefaktor.
3.3. Onkolytische Viren
Onkolytische Viren können Tumorzellen direkt oder indirekt zerstören. Im engeren Sinne
infizieren onkolytische Viren die Tumorzelle und lysieren diese. Es gibt aber weitere
Möglichkeiten, mittels Virus-basierter Methoden die Tumorzellen anzugreifen. Dazu
gehören das Einschleusen von Tumorsuppressorgenen und Toxinen in die Tumorzelle oder
die Generation einer Immunantwort mit darauffolgender Tumorzellzerstörung. Es werden
daher onkolytische Viren verwendet, die sowohl direkt einen Tumorzelltod verursachen
können, als auch die systemische Immunantwort stimulieren können [82]. Studien, die den Einsatz der onkolytischen Viren analysieren, werden bereits in
größeren Phase II- und III-Studien durchgeführt. In einer Phase III-Studie bei Patienten
mit fortgeschrittenem Malignen Melanom konnte mit einer Herpes simplex-basierten viralen
Therapie (talimogene laherparepvec, T-VEC) eine Verbesserung der Ansprechraten beobachtet
werden [83]. Außerdem werden zur Zeit Studien im Bereich des Hepatozellulären Karzinoms (Phase
II, pexastimogene devacirepvec, Posavec, NCT01387555) und des Kopf-Hals-Karzinoms
(Phase III, pelareorep, Reolysin in Kombination mit Chemotherapie, NCT01166542) durchgeführt
[84].
3.4. Monoklonale Antikörper
1975 entwickelten Kohler und Milstein eine Technologie, mit der es möglich ist, nahezu
unbegrenzte Mengen eines einzelnen, spezifischen Antikörpers herzustellen [85]. Durch die Verschmelzung einer Antikörper-produzierenden B-Zelle mit einer unsterblichen
„Myelom-Zelle“ entsteht ein ‚Hybridom’, das einen Antikörper einer Ursprungs-B-Zelle
produziert; man spricht von einem monoklonalen Antikörper. 1984 erhielten Kohler und
Milstein für diese Arbeiten zusammen mit Niels Jerne den Nobelpreis für Physiologie
oder Medizin. Dies würdigte insbesondere die Anwendungsmöglichkeiten, hochspezifische
Antikörper in theoretisch unbegrenzten Mengen herstellen zu können. Für die Anwendung
von Antikörpern in Forschung und Klinik war dies der Durchbruch, denn es standen nun
Antikörper in ausreichender Menge zur Verfügung, um etwa auf molekularer Ebene gezielt
in zelluläre Signalwege einzugreifen.
Für einen therapeutischen Einsatz waren monoklonale Antikörper anfangs nur beschränkt
geeignet. Da sie in der Regel aus Mäusen stammten, waren sie für den Menschen ‚fremd’
und induzierten eine Immunantwort, die sogenannte HAMA-Reaktion (human antibodies
against mouse antibodies). Mittlerweile lassen sich Antikörper herstellen, die teilweise
oder vollständig humanen Ursprungs sind. Die Terminologie der therapeutischen Antikörper
gibt Auskunft über Ursprung und Zusammensetzung: Die Endung -omab steht für Antikörper
murinen Ursprungs, -imab für Antikörper mit Ursprung von Primatenspezies, -ximab für
chimäre Antikörper (variabler Antikörperteil murin, Rest human), -zumab für humanisierte
Antikörper (Antigenbindungsstelle murin, Rest human) und -umab für vollständig humane
Antikörper ([Abb. 9]).
 Abb. 9 Aufbau der monoklonalen Antikörper. Monoklonale Antikörper werden abhängig von ihrer
Zusammensetzung und dem daraus resultierenden humanen Anteil unterschieden.
Abb. 9 Aufbau der monoklonalen Antikörper. Monoklonale Antikörper werden abhängig von ihrer
Zusammensetzung und dem daraus resultierenden humanen Anteil unterschieden.
Die therapeutische Spannbreite, in der mAb mittlerweile eingesetzt werden, ist groß.
Sie reicht von Autoimmunerkrankungen wie der rheumatoiden Arthritis über hämatologische
Erkrankungen wie der Hämophilie hin zu onkologischen Erkrankungen wie dem Malignen
Melanom, dem Bronchialkarzinom oder auch den Kopf-Hals-Karzinomen [86]. Die Onkologie stellt dabei ein Hauptgebiet in der Entwicklung, Erforschung und
Anwendung monoklonaler Antikörper dar, die meist gegen Tumorantigene oder Checkpoint-Rezeptoren
gerichtet sind.
3.4.1. mAb gegen Tumorantigene
Tumorantigene sind Proteinstrukturen, die von Tumorzellen produziert werden und eine
Immunantwort auslösen können. Man kennt tumorspezifische Antigene (TSA, engl. Tumor-Specific
Antigens) und tumorassoziierte Antigene (TAA, engl. Tumor-Associated Antigens). TSA
werden ausschließlich von Tumoren produziert und entstehen bspw. durch Mutationen
in einem Protein-kodierenden Gen. TAA dagegen kommen auch auf gesunden Zellen vor,
werden aber auf Tumorzellen deutlich höher exprimiert. Beispiele hierfür sind die
TAA der ErbB-Familie, eine Rezeptorfamilie von vier Tyrosinkinaserezeptoren. Zwei
Familienmitglieder, ErbB-1 (EGFR) und ErbB-2 (Her2), sind in der Onkologie von besonderer
Bedeutung. EGFR wird auf den meisten Kopf-Hals-Karzinomen, Nichtkleinzelligen Bronchialkarzinomen
(NSCLC, engl. Non Small Cell Lung Cancer) und kolorektalen Karzinomen überexprimiert,
Her2 in Mamma- und Ovarialkarzinomen [87]. Antikörper gegen EGFR und Her2 werden seit Jahren in der Klinik angewendet (Cetuximab,
Trastuzumab, [Tab. 1]) [88]. Leider liegen die Ansprechraten auf diese Therapien deutlich unter den Expressionsraten,
weshalb neue Entwicklungen auf Kombinationstherapien fokussieren, um Resistenzen der
Tumorzellen effektiver umgehen zu können.
Tab. 1 Tumorantigen-mAb.
|
Tumorantigen
|
mAb
|
Indikation
|
|
CA-125
|
Oregovomab
|
Ovarialkarzinom
|
|
CD19
|
Blinatumomab
|
Akute Lymphatische Leukämie (ALL)
|
|
CD20
|
Ofatumumab
|
Chronisch Lymphatische Leukämie (CLL)
|
|
CD20
|
Obinutuzumab
|
CLL, Non Hodgkin Lymphome (NHL)
|
|
CD20
|
Rituximab
|
NHL
|
|
CD20
|
Ibritumomab
|
NHL
|
|
CD20
|
Tositumomab
|
NHL
|
|
CD22
|
Inotuzumab
|
AML
|
|
CD22
|
Epratuzumab
|
NHL, ALL
|
|
CD33
|
Gemtuzumab
|
AML
|
|
CD38
|
Daratumumab
|
Multiples Myelom
|
|
CD4
|
Zanolimumab
|
T Zell-Lymphome
|
|
CD52
|
Alemtuzumab
|
ALL, CLL, T Zell-Lymphome
|
|
DLL3 (delta-like protein 3)
|
Rovalpituzumab
|
Kleinzelliges Bronchialkarzinom (SCLC)
|
|
EGFR
|
Necitumumab
|
Nichtkleinzelliges Bronchialkarzinom (NSCLC), Magenkarzinom
|
|
EGFR
|
Cetuximab
|
Kopf-Hals-Karzinome, Kolonkarzinome
|
|
EGFR
|
Panitumumab
|
Solide EGFR+ Tumoren
|
|
EpCAM-Antigen
|
Catumaxomab
|
maligner Aszites
|
|
HER2/neu-Rezeptor
|
Trastuzumab
|
Mammakarzinom, Magenkarzinom
|
|
HER2/neu, HER2/neu-Rezeptor
|
Pertuzumab
|
Ovarialkarzinom, Mammakarzinom, Bronchialkarzinom, Prostatakarzinom
|
3.4.2. mAb gegen Checkpoint-Moleküle
Eine neue Möglichkeit, spezifisch in die Signalwege des Immunsystems eingreifen zu
können, ergibt sich mit der Einführung der mAb, die Checkpoint-Moleküle stimulieren
oder blockieren können. Checkpoint-Moleküle sind überwiegend Rezeptoren auf Zellen
des Immunsystems, vorwiegend T-Lymphozyten, die eine stimulierende bzw. eine inhibierende
Wirkung ausüben.
Aus der Gruppe der Checkpoint-Moleküle sind die Moleküle CTLA4 und PD1 besonders hervorzuheben,
da mit therapeutischen Antikörpern, die gegen diese Moleküle gerichtet sind, in den
letzten Jahren teils signifikante therapeutische Erfolge in unterschiedlichen Tumorentitäten
erreicht werden konnten ([Tab. 2]). PD1 ist ein Rezeptor für die Liganden PDL1 und PDL2. Insbesondere die Bindung
von PDL1 führt zur T-Zell-Anergie ([Abb. 10]), d. h. die IFNγ-Sekretion und die Proliferation von T-Zellen werden supprimiert.
Im Tumormilieu wird dadurch eine Expansion aktivierter, Tumor-reaktiver T-Lymphozyten
verhindert [8]. Interessanterweise ist PD1 auch stark auf Treg exprimiert, führt bei diesen jedoch
zu einer Steigerung der Funktionalität [89]. Eine PD1-Blockade mit dem mAb Nivolumab wurde erstmals 2010 erfolgreich in einer
klinischen Phase I-Studie angewendet [90]. Seitdem konnten langanhaltende Erfolge erzielt werden mit vereinzelt kompletter,
über 3 Jahre anhaltender Regression von Patienten mit Malignem Melanom, Nierenzellkarzinom
oder Kolorektalkarzinom [91].
 Abb. 10 Wirkung der Checkpoint-Moleküle und der Checkpoint-Blockade. a Die CD8+ T-Zelle wird über die Bindung des PD1-Rezeptors durch das von den Tumorzellen
exprimierte PDL1 inhibiert. Die Bindung des Rezeptors führt zur T-Zell-Anergie durch
Hemmung der Proliferation und der T-Zell Funktion. b Durch die Checkpoint-Blockade wird die inhibierende Wirkung aufgehoben. TZR: T-Zell-Rezeptor-Komplex;
PD1: Programmed Death-Rezeptor 1; PDL1: PD1 Ligand.
Abb. 10 Wirkung der Checkpoint-Moleküle und der Checkpoint-Blockade. a Die CD8+ T-Zelle wird über die Bindung des PD1-Rezeptors durch das von den Tumorzellen
exprimierte PDL1 inhibiert. Die Bindung des Rezeptors führt zur T-Zell-Anergie durch
Hemmung der Proliferation und der T-Zell Funktion. b Durch die Checkpoint-Blockade wird die inhibierende Wirkung aufgehoben. TZR: T-Zell-Rezeptor-Komplex;
PD1: Programmed Death-Rezeptor 1; PDL1: PD1 Ligand.
Tab. 2 Checkpoint-Rezeptoren-mAb.
|
Checkpoint-Rezeptoren
|
mAb
|
Indikation
|
|
CTLA4
|
Tremelimumab
|
Bronchialkarzinom, Mesotheliom
|
|
CTLA4
|
Ipilimumab
|
Malignes Melanom
|
|
PD1
|
Nivolumab
|
Malignes Melanom Nichtkleinzelliges Bronchialkarzinom (NSCLC)
|
|
PD1
|
Pembrolizumab
|
Malignes Melanom, Mesotheliom, NSCLC
|
|
PDL1
|
Atezolizumab
|
Blasenkarzinom
|
|
PDL1
|
Avelumab
|
Blasenkarzinom, NSCLC, Merkelzellkarzinom
|
|
PDL1
|
Durvalumab
|
Bronchialkarzinom
|
CTLA4 wird auf T-Zellen exprimiert und induziert eine Immunsuppression über 2 wesentliche
Mechanismen: Einerseits agiert es aufgrund seiner strukturellen Ähnlichkeit zum ko-stimulatorischen
Molekül CD28 als kompetitiver Bindungspartner für die Oberflächenmoleküle CD80 und
CD86 auf antigenpräsentierenden Zellen und verhindert so die Aktivierung von T-Zellen
([Abb. 11]). Zum anderen inhibiert die Bindung von CTLA4 die T-Zellen direkt, unter anderem
durch Inaktivierung der T-Zell-Rezeptoren. Anti CTLA4-mAb waren die ersten Immuncheckpoint-mAb,
die klinisch getestet wurden [92] und signifikante, langanhaltende Therapieerfolge erzielten [2].
 Abb. 11 Wirkung der Checkpoint-Moleküle und der Checkpoint-Blockade. a Die CD4+ T-Zelle wird auf mehreren Ebenen inhibiert: Die Bindung des PD1 Rezeptors
durch PDL1 führt zur T-Zell-Anergie (1). Das fehlende ko-stimulatorische Signal durch
die Antigen-präsentierende Zelle bewirkt ebenso eine Inhibition (2). Das von der Treg
exprimierte CTLA4 bindet den ko-stimulatorischen Liganden der Antigenpräsentierenden
Zelle kompetitiv (3) und verhindert so das ko-stimulatorische Signal. b Durch die Checkpoint-Blockade wird die inhibierende Wirkung bei (1) aufgehoben. Ebenso
steht der ko-stimulatorische Ligand (CD80/86) wieder für das ko-stimulatorische Signal
zur Verfügung (2), da der kompetitive Bindungspartner CTLA4 ebenso durch einen Antikörper
blockiert ist (3). TZR: T-Zell-Rezeptor-Komplex; PD1: Programmed Death-Rezeptor 1;
PDL1: PD1 Ligand; CTLA4: Zytotoxische T-Lymphozyten-assoziiertes Protein; CD28: Ko-stimulatorischer
Rezeptor; CD80/86 Ko-stimulatorischer Ligand.
Abb. 11 Wirkung der Checkpoint-Moleküle und der Checkpoint-Blockade. a Die CD4+ T-Zelle wird auf mehreren Ebenen inhibiert: Die Bindung des PD1 Rezeptors
durch PDL1 führt zur T-Zell-Anergie (1). Das fehlende ko-stimulatorische Signal durch
die Antigen-präsentierende Zelle bewirkt ebenso eine Inhibition (2). Das von der Treg
exprimierte CTLA4 bindet den ko-stimulatorischen Liganden der Antigenpräsentierenden
Zelle kompetitiv (3) und verhindert so das ko-stimulatorische Signal. b Durch die Checkpoint-Blockade wird die inhibierende Wirkung bei (1) aufgehoben. Ebenso
steht der ko-stimulatorische Ligand (CD80/86) wieder für das ko-stimulatorische Signal
zur Verfügung (2), da der kompetitive Bindungspartner CTLA4 ebenso durch einen Antikörper
blockiert ist (3). TZR: T-Zell-Rezeptor-Komplex; PD1: Programmed Death-Rezeptor 1;
PDL1: PD1 Ligand; CTLA4: Zytotoxische T-Lymphozyten-assoziiertes Protein; CD28: Ko-stimulatorischer
Rezeptor; CD80/86 Ko-stimulatorischer Ligand.
3.4.2.1. Immunstimulierende Checkpoint-Moleküle Zu den immunstimulierenden Checkpoint-Molekülen zählen CD27, CD28, CD40, CD122, CD134
(OX40), CD137, CD278 (ICOS) und GITR (engl. Glucocorticoid-Induced TNFR family Related
Gen) ([Tab. 3]) [93]. CD27 ist wichtig für die Induktion von Gedächtnis T-Zellen und unterstützt die
T-Zell Expansion. CD28 ist ein wesentlicher ko-stimulierender T-Zell-Rezeptor, der
für die Aktivierung von CD4+ T-Zellen benötigt wird und mit CD80 und CD86 von antigenpräsentierenden
Zellen bindet. CD40 wiederum ist auf antigenpräsentierenden Zellen exprimiert und
wird durch seinen Liganden, CD40L, der auf CD4+ T-Zellen exprimiert wird, aktiviert.
CD122 wird auf CD8+ T-Zellen exprimiert und unterstützt deren Proliferation. CD134
(OX40) wird auf CD4+ und CD8+ T-Zellen exprimiert und wirkt gleichfalls proliferationsfördernd.
Zusätzlich verhindert eine Stimulation mittels OX40 die Ausbildung von Treg Zellen.
CD137 wird auf CD8+ T-Zellen exprimiert, stimuliert die Proliferation und verhindert
die aktivierungsabhängige Induktion einer Apoptose. CD278 (ICOS, engl. Inducible T
cell COStimulator) ist auf aktivierten T-Zellen exprimiert und interagiert mit antigenpräsentierenden
Zellen und B-Zellen. GITR stimuliert die Expansion von T-Zellen und wird ebenfalls
von antigenpräsentierenden Zellen stimuliert. Für viele der stimulierenden Checkpoint-Rezeptoren
werden aktuell klinische Studien durchgeführt, um über medikamentöse Liganden eine
verstärkte, anhaltende Immunantwort generieren zu können.
Tab. 3 Immunstimulierende Checkpoint-Moleküle.
|
Molekül
|
Exprimiert auf
|
Wirkung
|
|
CD27
|
(Gedächtnis-)T-Zellen
|
T-Zell-Expansion
|
|
CD28
|
CD4+/CD8+ T-Zellen
|
Essentielles Signal für T-Zell-Aktivierung
|
|
CD40
|
APC
|
Bindet mit CD40L auf T-Zellen, stimuliert Aktivität
|
|
CD122
|
CD8+
|
Proliferation
|
|
CD134 (OX40)
|
CD4+/CD8+ T-Zellen
|
Proliferation
|
|
CD137(4-1BB)
|
CD8+
|
Schutz vor Apoptose, Proliferation
|
|
CD278 (ICOS)
|
CD4+/CD8+ T-Zellen
|
Interaktion mit APC und B-Zellen
|
|
GITR
|
CD4+/CD8+ T-Zellen
|
Proliferation
|
3.4.2.2. Inhibierende Checkpoint-Moleküle Zu den inhibierenden Checkpoint-Molekülen die bisher identifiziert werden konnten,
gehören die Rezeptoren A2AR (Adenosin A2A Rezeptor), B7-H3, B7-H4, BTLA (engl. B and
T Lymphocyte Attenuator), CTLA4, KIR, LAG3, PD1, TIM3 (engl. T-cell Immunoglobulin
and Mucin domain 3) und VISTA (engl. V-domain Ig Suppressor of T cell Activation)
([Tab. 4]). A2AR vermittelt über die Adenosinbindung im Tumormilieu eine supprimierende Aktivität
auf Zellen des Immunsystems [94]. B7-H3 und B7-H4 werden auf Tumorzellen exprimiert, inhibieren T-Zellen und fördern
die Tumormigration [95]. BTLA wird auf T-Zellen exprimiert und führt nach Bindung zu einer Inhibition der
T-Zell Aktivität [96]. KIR werden insbesondere auf NK-Zellen exprimiert und können inhibierend auf die
NK-Zellfunktion wirken [97]. LAG3 wirkt insbesondere über die MHC II-Bindung immunsuppressiv auf CD4+ T-Zellen
und ist ein wichtiges Molekül in der supprimierenden Funktion von Treg Zellen [98]. TIM3 wiederum ist für die Aktivierung von Makrophagen verantwortlich, gleichzeitig
kann eine Apoptose von TH1-Zellen durch die Bindung von Galectin-9 induziert werden [99]. VISTA wurde als weiterer immunregulatorischer Checkpoint in therapierefraktären
Melanompatienten identifiziert [100].
Tab. 4 Immuninhibierende Checkpoint-Moleküle.
|
Molekül
|
Exprimiert auf
|
Wirkung
|
|
A2AR
|
T-Zellen
|
T-Zell-Inhibition, TGFβ Induktion
|
|
B7-H3
|
Tumorzellen
|
T-Zell-Inhibition, Tumormigration
|
|
B7-H4
|
Tumorzellen
|
T-Zell-Inhibition, Tumormigration
|
|
BTLA
|
T-Zellen
|
T-Zell-Inhibition
|
|
CTLA4
|
Treg, CD4+/CD8+ T-Zellen
|
T-Zell-Inhibition, APC Inhibition
|
|
KIR
|
NK-Zellen
|
NK-Zell-Inhibition
|
|
LAG3
|
Treg, CD4+/CD8+ T-Zellen
|
T-Zell-Inhibition
|
|
PD1
|
Treg, CD4+/CD8+ T-Zellen
|
T-Zell-Anergie von CD4+/CD8+ T-Zellen
|
|
TIM3
|
CD4+/CD8+ T-Zellen
|
T-Zell-Apoptose
|
|
VISTA
|
MDSC, Treg
|
T-Zell-Inhibition
|
Andere mAb sind gegen Zytokine oder Wachstumsfaktoren gerichtet ([Tab. 5]). Für viele der vorgestellten Moleküle sind die intra- und extrazellulären Signalwege
noch nicht vollständig entschlüsselt – mit entsprechend unentdeckten Wirkmechanismen.
Eine strikte Einteilung sollte daher mit Vorsicht erfolgen, da viele Signalwege in
der Immunologie sowohl stimulierende als auch inhibierende Effekte induzieren können.
Dies kann nicht nur zu teils widersprüchlich erscheinenden präklinischen Resultaten
führen, sondern auch ein unterschiedliches Therapieansprechen bewirken. Umso wichtiger
ist es daher, bereits in präklinischen Studien die Wirkweise der Signalwege genau
zu analysieren und kritisch in vitro-Ergebnisse mit validen in vivo-Modellen zu überprüfen.
Tab. 5 Sonstige onkotherapeutisch eingesetzte mAb.
|
Zielstruktur
|
mAb
|
Indikation
|
|
CCR4
|
Mogamulizumab
|
Adulte T-Zell-Leukämie, NHL
|
|
HLA-DR
|
Apolizumab
|
Akute lymphatische Leukämie (ALL), Chronisch lymphatische Leukämie (CLL), Non Hodgkin
Lymphome (NHL), solide Tumoren
|
|
IgG1 auf PDGF Rezeptor-α (platelet-derived growth factor receptor α)
|
Olaratumab
|
Sarkom
|
|
IL6-Rezeptor
|
Tocilizumab
|
Zytokinsturm nach CART-Zelltherapie
|
|
IL6
|
Siltuximab
|
Multiples Myelom
|
|
RANK Ligand (Rezeptoraktivator des NF-κB Liganden, RANKL)
|
Denosumab
|
Knochenmetastasen
|
|
Signaling Lymphocyte Activation Molecule (SLAMF7)
|
Elotuzumab
|
Multiples Myelom
|
|
VEGF (Vascular Endothelial Growth Factor)
|
Bevacizumab
|
Kolonkarzinom, Mammakarzinom, Nichtkleinzelliges Bronchialkarzinom (NSCLC)
|
|
VEGF Rezeptor
|
Ramucirumab
|
Bronchialkarzinom, Magenkarzinom
|
3.5. Vakzinierung
In der Onkologie müssen 2 Vakzinierungsstrategien unterschieden werden: Zum einen
kann eine präventive Vakzinierung vor der Entstehung von Krebs angewendet werden.
Dies ist in Zusammenhang mit Impfungen gegen onkogene Viren möglich und kann daher
einen protektiven Effekt gegen viral assoziierte Karzinome bewirken. Zum anderen gibt
es Bestrebungen, in anderen, i. e. nicht ausschließlich viral bedingten Tumoren, eine
therapeutische Vakzinierung zu etablieren.
Präventive Impfungen zur Vermeidung von Infektionen mit onkogenen Viren haben sich
als hocheffizient in der Vermeidung von virus-assoziierten Karzinomen gezeigt. Durch
die Entwicklung von Vakzinierungen gegen die Hochrisiko-Virussubtypen der onkogenen
humanen Papillomaviren (HPV) 16 und 18 konnte eine Protektion vor HPV-Infektionen
erreicht werden [101]. Für die Arbeiten, die zur Entdeckung des Zusammenhangs zwischen HPV-Infektionen
und dem Zervixkarzinom geführt hatten, erhielt Harald zur Hausen 2008 den Nobelpreis
für Medizin oder Physiologie [102]. Daher besteht mittlerweile eine Impfempfehlung für die Hochrisiko-Virussubtypen
für junge Frauen, möglichst vor dem ersten Geschlechtsverkehr. Mittlerweile ist bekannt,
dass onkogene HPV-Infektionen in der westlichen Zivilisation nicht nur für fast alle
Zervixkarzinome, sondern auch für über 90% der Analkarzinome, 70% der Oropharynxkarzinome,
70% der Vaginalkarzinome, 40% der Vulvakarzinome und 50% der Peniskarzinome verantwortlich
sind [101]. Aufgrund der hohen Inzidenz von HPV16-positiven Oropharynxkarzinomen wird untersucht,
ob eine Vakzinierung einen protektiven Effekt im Hinblick auf die Inzidenz von Kopf-Hals-Karzinomen
bewirken kann [103]. Daher wird auch eine Impfempfehlung für Jungen und junge Männer diskutiert [104]. Der Einfluss der in den letzten Jahren begonnenen Impfungen für Mädchen und junge
Frauen auf diese epidemiologischen Zahlen wird in den kommenden Jahren besser zu analysieren
sein.
Im Gegensatz zu präventiven Impfungen, die gegen onkogene virale Strukturen immunisieren,
sind auch therapeutische Impfungen gegen Tumorzellantigene in der Entwicklung. Anders
als in der Therapie mit mAb soll hier jedoch das Immunsystem selbst gegen die Tumorantigene
aktiviert werden, um so spezifische T-Zellen, Antikörper und andere Komponenten insbesondere
der adaptiven Immunantwort gegen die Tumorzellen zu mobilisieren. Diese Aktivierung
basiert auf dem Zusammenspiel zwischen Dendritischen Zellen und T-Zellen. Hierzu existieren
verschiedene Techniken, wie Dendritische Zellen mit den Tumorantigenen konfrontiert
werden: Entweder werden die DC mit Tumorlysaten, Proteinen oder Peptiden beladen oder
mit DNA oder RNA transfiziert [105].
Aktive onkologisch wirksame Vakzinierungen sind in der Klinik bisher noch nicht breit
etabliert. Gerade bei in situ-Karzinomen konnten jedoch in klinischen Studien substantielle
Erfolge erzielt werden. Bei intraepithelialen Neoplasien, z. B. bei der vulvären intraepithelialen
Neoplasie (VIN), führten Impfungen gegen HPV16 Onkoproteine zu einem Regress der Läsionen,
wobei das Therapieansprechen unmittelbar mit der durch die Impfung induzierten T-Zellantwort
korrelierte [106]
[107]
[108]. Ähnliche Ergebnisse zeigten Czerniecki und Kollegen beim Duktalen Karzinom in situ
(DCIS) mit einer gegen Her2/neu gerichteten Impfung [109]. Morse und Kollegen belegten ein signifikant erhöhtes Gesamtüberleben bei Patienten
mit resezierten Kolonkarzinom-Metastasen nach einer Impfung gegen die Tumorantigene
CEA und MUC1[110]. In einer Phase I-Studie mit einer Vakzinierung gegen das Tumorantigen p53 konnte
bei Kopf-Hals-Karzinompatienten eine 2-Jahresüberlebensrate von 88% beobachtet werden
[111].
Eine Schwierigkeit, die auch die Effektivität der Vakzinierungen beeinflussen kann,
stellt dabei das immunsuppressive Tumormilieu dar. Durch die Ausbildung von Treg,
TAM und MDSC werden immunsupprimierende Signalwege gefördert, die eine Expansion Tumorantigen-spezifischer
T-Zellen erschweren. Um dies zu vermeiden, beschreiben Mould und Kollegen bspw. eine
therapeutische Effizienzsteigerung durch multiple Impflokalisationen [112]. Andere Strategien zielen auf die Kombination mit anderen Immunstimulantien ab,
bspw. mit Toll-like-Rezeptor Agonisten [113].
3.6. Adoptiver Zelltransfer
Beim adoptiven Zelltransfer werden Lymphozyten (meist T-Zellen, aber auch Dendritische
Zellen, NK-Zellen u. a.) aus dem peripheren Blut der Patienten isoliert. Anschließend
können Tumorantigen-spezifische (T) Zellen hergestellt bzw. expandiert werden, die
dann dem Patienten wieder verabreicht werden [114]
[115] ([Abb. 12]). Ein Vorteil des adoptiven Zelltransfers besteht darin, die Lymphozyten außerhalb
des immunsuppressiven Tumormilieus beeinflussen und expandieren zu können [115].
 Abb. 12 Adoptiver Zelltransfer. (1) Lymphozyten werden dem Patienten entnommen, entweder
aus dem peripheren Blut, oder aus dem Tumor selbst. (2) Anschließend werden die Lymphozyten
isoliert und gegebenenfalls modifiziert. (3) Dann erfolgt die Selektion der Lymphozyten
mit der höchsten Spezifität. (4) Die selektierten Lymphozyten werden vermehrt und
anschließend reinfundiert (5).
Abb. 12 Adoptiver Zelltransfer. (1) Lymphozyten werden dem Patienten entnommen, entweder
aus dem peripheren Blut, oder aus dem Tumor selbst. (2) Anschließend werden die Lymphozyten
isoliert und gegebenenfalls modifiziert. (3) Dann erfolgt die Selektion der Lymphozyten
mit der höchsten Spezifität. (4) Die selektierten Lymphozyten werden vermehrt und
anschließend reinfundiert (5).
Erste klinische Studien zum adoptiven T-Zelltransfer wurden bereits in den 90er Jahren
durchgeführt [61]. Hier konnte in ca. 30% der behandelten Melanompatienten ein Therapieansprechen
erzielt werden. Mittlerweile werden in speziellen Tumorentitäten Remissionsraten von
bis zu 90% der behandelten Patienten berichtet, so z. B. bei Patienten mit akuten
CD19+ Leukämien [116]. Einen Beitrag dazu leistet die „Präkonditionierung“, bei der Patienten mit einer
Lymphodepletionstherapie vorbehandelt werden [117]. Dadurch werden (auch) suppressive Immunzellen wie regulatorische T-Zellen oder
MDSC reduziert, die die Effektivität der re-infundierten antigenspezifischen T-Zellen
inhibieren würden. Für eine erfolgreiche Antigen-spezifische Expansion der Zellen
werden Antigenpräsentierende Zellen benötigt. Zu den APC, die beim adoptiven Zelltransfer
für die Antigenpräsentation verwendet werden, gehören natürliche Dendritische Zellen,
künstliche Zellen oder mit Antigenen beladenen „Beads“. T-Zellen können aus dem peripheren
Blut oder aus dem Tumor gewonnen werden. Ex vivo erfolgt dann mit verschiedenen Methoden
eine Expansion der Tumorantigen-spezifischen Zellen auf eine Zellzahl von über 109-1011 T-Zellen [118]. Eine Selektion der Tumorantigen-spezifischen T-Zellen erfolgt nach Isolation einzelner
T-Zelllinien, die anschließend auf die Reaktivität gegen verschiedene Tumorantigene
getestet werden. So werden die T-Zelllinien expandiert, die die stärkste Reaktivität
auf die dargebotenen Tumorantigene aufweisen. Moderne Methoden bedienen sich einer
gentechnisch bestimmten Antigen-Spezifität der T-Zellen und können so noch präziser
und variabler auf spezifische Tumorantigene angepasst werden. Neben dem Vorteil extrakorporal
Antigen-spezifische T-Zellen selektiv auszuwählen und in großer Anzahl zu expandieren,
besteht diese Expansion außerdem nicht unter dem Einfluss des immunsuppressiven Tumormilieus.
Die T-Zellen können daher in einem funktionellen Zustand expandiert werden. Dabei
muss eine qualitativ hochwertige Expansion der T-Zellen gewährleistet werden, um die
Tumorantigen-Spezifität der T-Zellen zu erhalten. So ist nicht nur der Differenzierungsstatus
der T-Zellen wichtig, sondern auch die zellulären metabolischen Prozesse [119]. Um die re-infundierten, expandierten T-Zellen zu stimulieren, werden Zytokine verwendet
[120].
Wenn statt eines Tumorantigen-spezifischen T-Zell-Rezeptors ein chimärer Antigenrezeptor
(CAR) verwendet wird, spricht man von CAR-T-Zelltransfer. Die chimären Antigenrezeptoren
bestehen dabei aus einer antigenbindenden Komponente – bspw. einem Antikörper – sowie
einer weiteren, T-Zell-aktivierenden, ko-stimulierenden Komponente, um die Effektivität
der T-Zellantwort zu erhöhen [121]. Diese ko-stimulierende Komponente wirkt sich auf die Zytokinsekretion aus und kann
die Proliferation der T-Zellen stimulieren. In CAR-T-Zellen der dritten Generation
sind 2 oder mehr immunstimulierende Domänen in den Rezeptor integriert. Zu den verwendeten
ko-stimulatorischen Molekülen gehören CD28, OX-40 (CD134) oder 4-1BB (CD137). Besondere
Effektivität dieser therapeutischen Methoden konnte bisher bei hämatoonkologischen
Erkrankungen demonstriert werden. Gerade in B-Zell Malignomen zeigten sich eine gute
Ansprechrate und klinische Effektivität (NCT02435849, [116]
[122]). In soliden Tumoren stehen überzeugende Resultate in diesen Dimensionen noch aus.
Dies wird v. a. der Tatsache zugeschrieben, dass die Identifikation der spezifischen
Tumorantigene erschwert ist. Bisher wurden Studien zu CAR-T-Zelltherapien insbesondere
für Her2+ Tumoren, EGFR+ Tumoren, CEA+ (Karzinoembryonales Antigen, engl. CarcinoEmbryonic
Antigen) Tumoren und Mesothelin+ Tumoren durchgeführt [123]
[124]
[125]
[126].
4. Chancen/ Risiken
Eine wesentliche Aufgabe ärztlicher Tätigkeit ist es, den potentiellen Nutzen und
das Risiko einer therapeutischen Maßnahme gegeneinander abzuwägen. Dies gilt insbesondere
auf onkologischem Gebiet, da hier therapeutische Maßnahmen die Lebensqualität der
Patienten stark einschränken können. Daher ist es wichtig, auch die Nebenwirkungen
und möglichen Risiken der verschiedenen immuntherapeutischen Verfahren zu berücksichtigen.
Da das immuntherapeutische Prinzip darauf basiert, das Immunsystem zu stimulieren
und blockierende Auswirkungen des Tumormilieus aufzuheben, werden viele Nebenwirkungen
durch überschießende, autoimmune Prozesse hervorgerufen. Üblicherweise hängt dabei
das Nebenwirkungsprofil stark von der Breite des immuntherapeutischen Ansatzes ab.
Je pluripotenter die beeinflussten Signalwege, desto größer das Risiko möglicher Nebenwirkungen.
Dies gilt insbesondere für Zytokin-basierte Immuntherapien. So kann IL2 Nebenwirkungen
wie Übelkeit, Erbrechen, gastrointestinale Beschwerden, ein schweres Krankheitsgefühl,
erhöhte Kapillarpermeabilität, kardiale Schädigungen und einen niedrigen Blutdruck
verursachen [61]. Viele dieser Nebenwirkungen können so stark ausgeprägt sein, dass sie einen Therapieabbruch
erzwingen. Für IL21 wurden ähnliche Nebenwirkungen beschrieben, insbesondere Fieber,
Schüttelfrost, Leberschädigungen und Auswirkungen auf das hämatopoetische System mit
Neutropenie und Thrombozytopenie [127]. Interferone können neben einer Leukopenie Schwindel, Anorexie und Depressionen
induzieren [68].
Bei den monoklonalen Antikörpern, die gegen Tumorantigene gerichtet sind, beeinflussen
insbesondere 2 Aspekte die Ausbildung und das Profil der Nebenwirkungen: Zum einen
die zuvor beschriebene Zusammensetzung des Antikörpers. Mit steigendem „humanen“ Anteil
wird die Fremdreaktion des Körpers reduziert, eine allergische Reaktion gegen das
Medikament vermieden. Mittlerweile kann dies durch molekularbiologische Veränderung
der Antikörper erreicht werden. Zum anderen ist auch wichtig, ob das Zielmolekül des
Antikörpers auch auf gesunden Zellen vorkommt. Je unspezifischer das Tumorantigen
ist, desto stärkere Nebenwirkungen können beobachtet werden. Zusätzlich kann bei einer
hohen Tumorlast eine unkontrollierte Freisetzung von Zytokinen durch Zytolyse erfolgen
(engl. Cytokine-release Syndrome). Gegen Tumorantigen-gerichtete mAb können Fieber,
Schüttelfrost, Atembeschwerden und Hautausschläge verursachen. Unter der Behandlung
von Rituximab (anti-CD20 mAb) kann in seltenen Fällen eine progressive multifokale
Leukenzephalopathie (PML) auftreten [128]. Eine typische Nebenwirkung, die der anti-EGFR mAb Cetuximab hervorrufen kann, ist
ein Hautausschlag, dessen Ätiologie immer noch nicht vollständig geklärt ist [129]. Der anti-Her2 mAb Trastuzumab kann insbesondere kardiale Nebenwirkungen hervorrufen,
die eine regelmäßige Kontrolle der kardialen Funktion während und nach der Therapie
erfordern [130]. Bevacizumab wiederum ruft häufig gastrointestinale Beschwerden hervor, ebenso wie
eine arterielle Hypertonie und eine Proteinurie [131].
MAb, die gegen Checkpoint-Moleküle gerichtet sind, steigern die Immunreaktion. Dadurch
können Entzündungsreaktionen im Körper hervorgerufen werden. Nivolumab, ein anti-PD1
mAb, kann unter anderem eine Arthritis, eine Kolitis und insbesondere eine Pneumonitis
hervorrufen. Ipilimumab, ein anti-CTLA4 mAb, ist mit Nebenwirkungen assoziiert, die
unter anderem die Haut, Leber, Augen, Darm und Hypophyse betreffen.
Auch bei Vakzinierungstherapien können Impfreaktionen und andere Nebenwirkungen hervorgerufen
werden. Selbst die spezifische T-Zell-Rezeptorselektion im Rahmen des adoptiven T-Zelltransfers
kann Antigen-abhängig starke Nebenwirkungen verursachen. Daher werden aktuell Methoden
entwickelt, die die Wahrscheinlichkeit derartiger Komplikationen reduzieren. Kunert
und Kollegen schlagen spezifische Methoden vor, um eine Risiko-Einschätzung hinsichtlich
der Toxizität mittels TZR-Selektion zu ermöglichen [132]. Dazu gehört unter anderem die Analyse großer Genomdatenbanken, ob die Zielantigene
auch in anderen gesunden Geweben und Organen vorkommen. Ein anderer Ansatz liegt in
der Entwicklung von Kombinationstherapien, die eine synergistische, therapeutische
Wirkung ermöglichen, bei gleichzeitiger Reduktion der Einzeldosis und damit verbundener
dosisabhängiger Nebenwirkungen.
5. Aktuelle Entwicklungen
5. Aktuelle Entwicklungen
Eine herausragende klinische Relevanz im Bereich der Immuntherapien hatten in den
letzten Jahren insbesondere die Checkpoint-Inhibitoren. Bisher sind CTLA4-, PD1- und
PDL1-Inhibitoren in der onkologischen Therapie zugelassen. Der anti-CTLA4 mAb Ipilimumab
(Yervoy®, Bristol-Myers Squibb) ist seit 2011 in Europa bei der Behandlung des fortgeschrittenen,
metastasierten oder nicht resektablen Malignen Melanoms zugelassen. PD1-Inhibitoren
sind seit 2015 für mehrere Tumorentitäten erhältlich: Zur Behandlung des fortgeschrittenen
metastasierten oder nicht resektablen Malignen Melanoms. Als Second-Line-Therapie
besteht zusätzlich die Zulassung für die Behandlung des fortgeschrittenen Nichtkleinzelligen
Bronchialkarzinoms, des fortgeschrittenen Nierenzellkarzinoms und des fortgeschrittenen
Hodgkin Lymphoms. Außerdem stehen das therapierefraktäre Kopf-Hals-Karzinom und das
Urothelkarzinom kurz vor der Zulassung zur Indikation einer anti-PD1 Therapie. Bisher
zugelassene anti-PD1 mAb sind Nivolumab (Opdivo®, Bristol-Myers Squibb) und Pembrolizumab (Keytruda®, Merck Sharp & Dohme). Im März 2017 wurde der anti-PDL1 mAb Avelumab (Bavenico®, Merck KGaA, Pfizer und Eli Lilly and Company) für die Behandlung des metastasierten
Merkelzellkarzinoms durch die FDA in den USA zugelassen. Atezolizumab (Tecentriq®, Genentech/Roche), ebenfalls ein PDL1 mAb, ist seit 2016 in den USA zur Behandlung
für das fortgeschrittene oder metastasierte Urothelkarzinom und das vorbehandelte,
metastasierte Nichtkleinzellige Lungenkarzinom zugelassen. Anti-PDL1 mAb sind bisher
in Europa noch nicht zugelassen. Die Anzahl klinischer Studien, die zu Checkpoint-Modulatoren
durchgeführt werden, ist immens. Alleine für Kopf-Hals-Karzinome wurden seit 2010
über 45 klinische Phase I – III Studien durchgeführt [133]. Die Mehrzahl der Studien untersucht Medikamente, die den PD1/PDL1 Signalweg beeinflussen.
Aber auch andere immunstimulierende und -inhibierende Checkpoint-Moleküle werden erforscht.
Ein mAb zur Stimulation von OX40 zeigte in einer Phase I-Studie eine Regression metastatischer
Befunde in einem Drittel der Patienten (NCT01644968, [134]). Auch andere stimulatorische Moleküle wie CD137 oder inhibitorische Moleküle wie
LAG3 sind Gegenstand klinischer Studien (NCT02658981).
5.1. Biomarker
Nicht alle Patienten profitieren von den neu etablierten Therapien, die Gründe hierfür
sind meist unbekannt. Die zuvor erwähnten Nebenwirkungen und die hohen Therapiekosten
fordern daher die Identifikation von immuntherapeutischen prädiktiven Biomarkern,
die eine Prognose über das therapeutische Ansprechen und damit eine Therapieselektion
ermöglichen. Untersuchungen bisher zeigen hier jedoch ein heterogenes Bild. Insbesondere
die PDL1 Expression wurde in verschiedenen Studien untersucht, mit teils widersprüchlichen
Ergebnissen. Dies wird unter anderem darauf zurückgeführt, dass unterschiedliche Standards
zur PDL1 Bestimmung bestehen. Zusätzlich werden unterschiedliche Grenzwerte für eine
Positivität berücksichtigt. Außerdem ist die PDL1 Expression als Biomarker auch Tumor-abhängig.
So ist bei Nichtkleinzelligen Bronchialkarzinomen die PDL1 Expression als Biomarker
eher etabliert als bspw. bei Kopf-Hals-Karzinomen [135]. Auch bei PDL1 negativen Patienten wurde ein Therapieansprechen nach Blockade des
PD1 / PDL1 Signalweges beobachtet [136]. Daher fehlen bisher im Bereich der Checkpoint-Immuntherapie verlässliche Biomarker.
Kürzlich publizierte Untersuchungen an Patienten mit Kopf-Hals-Karzinomen weisen darauf
hin, dass das Ausmaß der PD1-Expression von CD8+ T-Zellen mit der Zellfunktionalität
und dem Gesamtüberleben der Patienten assoziiert ist [137].
Die Ansprechraten in der Immuntherapie hängen damit nicht nur von den Medikamenten
und den Tumorentitäten, sondern auch von den Patienten ab. Da die Ansprechraten variieren
und bei Monotherapien weiterhin meist unter 50% liegen, werden verstärkt Kombinationstherapien
eingesetzt, um bei möglichst vielen Patienten einen therapeutischen Erfolg erzielen
zu können. Dies erhöht zum einen die Chance, dass patientenspezifisch relevante Signalwege
beeinflusst werden können. Zum anderen können dadurch potentiell synergistische Therapieeffekte
erzielt werden.
5.2. Kombinationstherapien
Gerade die Checkpoint-Rezeptor-Therapien sind für eine Kombinationstherapie besonders
interessant, da sie direkt im Bereich der antitumorspezifischen Zellen ansetzen und
einen Mechanismus blockieren, der die Effektivität anderer Therapieansätze potentiell
behindert. Dies gilt sowohl für klassische therapeutische Verfahren wie die Strahlentherapie,
die nachweislich eine PDL1-Expression induziert [138], als auch für andere immuntherapeutische Ansätze. Auch in der Kombination mit Zytokintherapien
und Vakzinierungsstrategien kommen die Checkpoint-Rezeptor-Therapien zum Einsatz.
In präklinischen Studien wurde bspw. die Kombination eines GM-CSF modifizierten Prostatakarzinom-Vakzins
mit einer PDL1 Blockade untersucht [139], da bisherige Vakzinierungstherapien durch die PDL1 Induktion in ihrer Effektivität
supprimiert wurden. In dieser murinen Studie konnte ein starker Anstieg an CD4+ und
CD8+ T-Zellen mit gesteigerter IFNγ-Sekretion beobachtet werden, assoziiert mit einem
Tumorregress. Ein ähnliches Ergebnis konnte mit dem onkolytischen Virus Onkovex in
Kombination mit GM-CSF und anti-CTLA4 erreicht werden [140]. In diesem Modell konnten sowohl eine systemische Wirkung durch Behandlungserfolge
in nicht direkt injizierten Tumoren (50%), als auch eine lokale, höhere Effektivität
(100%) in direkt injizierten Tumoren beobachtet werden. Zusätzlich zur direkten onkolytischen
Therapie im Tumor war eine CD8+ T-Zellreaktion im nicht injizierten Tumor zu beobachten.
In einer klinischen Studie untersuchten Hodi und Kollegen die Kombination von GM-CSF
(Sargramostim) mit dem anti-CTLA4 mAb Ipilimumab in Patienten mit nicht resektablem
Malignen Melanom im Stadium III oder IV [141] und verglichen diese Therapie mit einer Ipilimumab Monotherapie. Im Rahmen dieser
Studie konnte kein signifikanter Unterschied im Hinblick auf das progressionsfreie
Überleben beobachtet werden, aber eine signifikante Verlängerung des Gesamtüberlebens
und eine Reduktion der Nebenwirkungen durch die Kombinationstherapie.
Zusammen mit den Checkpoint-Molekülen werden aktuell hohe Erwartungen an die adoptive
T-Zelltherapie gestellt. Aufgrund vielversprechender Ergebnisse ist die erste CAR-T-Zelltherapie
in den USA im August 2017 zugelassen worden. CTL019 (Tisagenlecleucel®, Novartis) wird damit zur Behandlung von Kindern und Jugendlichen mit akuter, therapierefraktärer
B-Zell Leukämie verwendet. Für die Zukunft wird ein deutlicher Anstieg der Zulassungen
und der damit verbundenen Indikationen erwartet. Eine Kombination der adoptiven T-Zelltherapie
mit Eingriffen in den PD1 Signalweg der CAR T-Zellen wird aktuell in einer Phase I
Studie an Patienten mit metastasiertem Nichtkleinzelligem Bronchialkarzinom untersucht
(NCT02793856). Hier wird durch die moderne Technik der CRISPR/Cas-Methode das Genom
der T-Zellen gezielt und exakt verändert und die PD1 Expression ausgeschaltet. Die
derart veränderten T-Zellen werden nach Selektion und Proliferation anschließend reinfundiert.
6. Zusammenfassung und Ausblick
6. Zusammenfassung und Ausblick
Die Interaktionen im Tumormilieu sind hochkomplex und stellen onkologische Therapieansätze
vor eine große Herausforderung. Trotzdem hat die onkologische Immuntherapie in den
letzten Jahren wesentliche Fortschritte erzielt. In einigen Tumorentitäten konnten
Erfolge mit langanhaltenden Therapieansprechraten, teilweise kompletten Remissionen
erzielt werden. Doch nicht alle Patienten können von den Entwicklungen bisher profitieren.
Bei vielen Tumorentitäten sind die Ansprechraten bei einem monotherapeutischen Ansatz
begrenzt. Dennoch bilden Fortschritte in der Grundlagenforschung und der klinischen
Forschung die Basis für ein verbessertes Verständnis der Interaktion zwischen Tumor
und Immunsystem. Dies ermöglicht neue therapeutische Kombinationen, um synergistische
Effekte zu nutzen. Insbesondere die Entwicklungen der Checkpoint-spezifischen Antikörper
hebt die Blockade durch immuninhibierende Signalwege auf und lässt Kombinationen mit
anderen therapeutischen Strategien, die bisher aufgrund dieser Inhibition nicht von
Erfolg gekrönt waren, vielversprechend erscheinen. Gleichzeitig können immunstimulierende
Signalwege Zellen stärken, die bisher durch das Tumormilieu supprimiert wurden, und
zu einer Verbesserung der Immunantwort führen. Die Etablierung prädiktiver Marker
und eine damit verbundene Verbesserung der Patientenselektion für die verschiedenen
Therapiemodalitäten wird zukünftig weiter an Bedeutung gewinnen. Gerade weil die Immuntherapie
in der Onkologie ein derartiges Potenzial aufweist, ist aber auch ein kritischer und
reflektierter Umgang mit Studienergebnissen und therapeutischen Ansätzen wichtig,
um der aktuellen und zukünftigen Bedeutung im Klinikalltag gerecht zu werden.
Anmerkung: Die Abbildungen des Beitrags wurden mit Microsoft Powerpoint 2016© (Microsoft Corporation, Redmond, USA) und Science Slides© (VisiScience Inc., Chapel Hill, USA) erstellt.





