

Subscribe to RSS
DOI: 10.1055/s-0043-1772821
Thiamine-Responsive Megaloblastic Anemia Syndrome Combined with Thalassemia Trait: A Rare Association
Authors
Funding and Sponsorship None.

Abstract
Introduction Thiamine-responsive megaloblastic anemia syndrome (TRMA, OMIM reference 249270), also known as Rogers' syndrome, is a rare type of anemia characterized by the triad megaloblastic anemia, sensorineural hearing loss, and diabetes mellitus (DM). Disturbance of thiamine transport into cells results from homozygous or compound heterozygous mutations in the SLC19A2 gene.
Case Report We report the case of an 8-year-old girl who presented at age 4 years with anemia. She had a combined hematological profile of microcytic and macrocytic anemia. The parents refused bone marrow aspiration and genetic diagnosis. Hemoglobin electrophoresis established the thalassemia trait. She was later confirmed to have sensorineural deafness and monogenic DM. A tentative TRMA diagnosis was based on megaloblastic anemia, sensorineural deafness, and monogenic DM triad. The patient was treated empirically with a daily dose of thiamine 200 mg; her hemoglobin level normalized, but the deafness and DM did not improve.
Conclusion In routine practice, patients with TRMA must be evaluated thoroughly for other causes of megaloblastic anemia, including therapeutic thiamine trials in the presence of sensorineural deafness or DM. These patients should be followed throughout their life span both for DM and to control their response to thiamine therapy for megaloblastic anemia.
Keywords
monogenic diabetes mellitus - sensorineural deafness - megaloblastic anemia - vitamin B1 deficiency - thalassemia traitIntroduction
Thiamine-responsive megaloblastic anemia (TRMA), or Rogers' syndrome, is an autosomal recessive condition initially described in 1969. Although several clinical findings are sometimes presented simultaneously, it is classically characterized by the development of diabetes mellitus (DM), megaloblastic anemia, and sensorineural deafness.[1] It is caused by intracellular thiamine deficiency, resulting from defective thiamine transporter protein 1 (THTR1).[2] Monogenic DM is a rare DM type due to a single gene that causes early onset pancreatic β-cell damage.[3]
TRMA generally responds to thiamine therapy, in varying degrees, if started in fetal or early life. Approximately 40 cases have been reported to date, and it is considered extremely rare outside consanguineous marriage. Cases have been documented in patients of almost all racial and ethnic origins. TRMA diagnosis is commonly established by whole-exome SLC19A2 gene sequencing.[4] This gene encodes the thiamine transporter protein found in the cell membranes of various tissue types, and while tissues typically have two of these transporters, the β cells of the pancreas, the bone marrow, and a subgroup of cochlear hair cells have only one. As such, a mutation in this gene leads to a deficiency in thiamine, vitamin B1, and is clearly and directly related to the development of the trio mentioned earlier of characteristic conditions that classically present with TRMA.[5]
Patients and Methods
The patient was an 8-year-old girl who was the offspring of a consanguineous marriage between Arab parents who were first-degree cousins. She was born full term after a normal delivery, with a birthweight of 2.7 kg and a normal systemic physical examination. On day 1, she was admitted to a neonatal intensive care unit for 1 week because of respiratory distress and neonatal sepsis, for which she received a full course of antibiotics. There was no relevant family history other than maternal gestational diabetes. Specifically, there was no family history of anemia. The patient presented with recurrent anemia during infancy and early childhood and was subsequently treated with iron and vitamin supplements. She was 3.6 years old when her parents relocated to the United Arab Emirates from another Arab country.
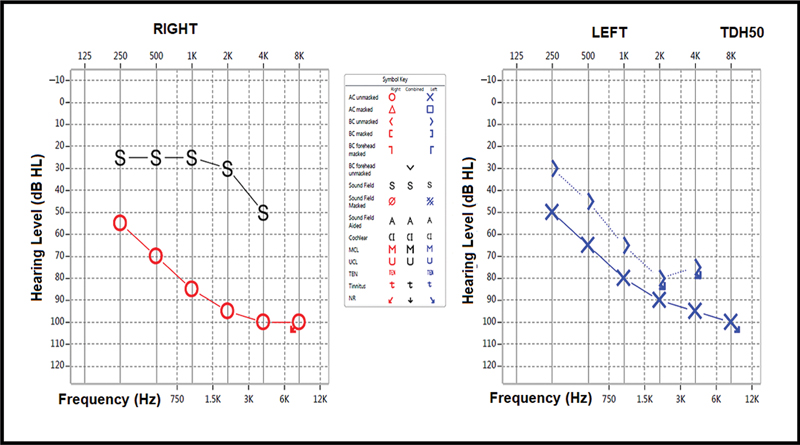
At age 4 years, she presented with symptoms, signs, and biochemical markers of diabetic ketoacidosis. Her blood glucose was 30 mmol/L, and serum insulin was undetectable. Antibodies to glutamic acid decarboxylase, antityrosine phosphatase, and anti-insulin antibodies were reported as negative. Insulin therapy was started based on type 1 DM diagnosis. Her hemoglobin was 5.5 g/dL, and the blood film showed a mixed picture of hypochromic microcytic red blood cells (RBCs) with a moderate number of macrocytes. The parents refused the recommended bone marrow aspiration procedure. Other investigations ruled out iron, folate, and vitamin B12 deficiencies. Hemoglobin electrophoresis ([Table 1]) showed the β-thalassemia trait without evidence of hemolysis. After receiving a blood transfusion, her hemoglobin increased to 10.5 mg/dL. The audiogram showed profound bilateral sensorineural deafness ([Fig. 1]).
Abbreviations: HbA, adult hemoglobin; HbF, fetal hemoglobin; HPLC, high-performance liquid chromatography; RBC, red blood cell.
Electrocardiogram (ECG) showed normal sinus rhythm with ventricular bigeminy corresponding to Purkinje's cell premature ventricular contractions (PVCs), mainly at the left posterior fascicle. The PR interval and QRS duration were normal ([Fig. 2]). Arrhythmias and PVCs had never been reported previously but were an incidental finding. Transthoracic Doppler echocardiography showed normal ejection fraction with no evidence of cardiomyopathy. She had no symptoms or signs of cerebral involvement, and brain magnetic resonance imaging was normal.

In summary, this patient was diagnosed with monogenic DM, macrocytic anemia, sensorineural hearing loss, and thalassemia trait. Given these findings, the diagnosis of TRMA with thalassemia trait was considered. The parents did not give their consent for genetic testing. Thiamine treatment at 200 mg was prescribed, and insulin therapy via an insulin pump was commenced. She initially had hearing aids and later received cochlear implants.
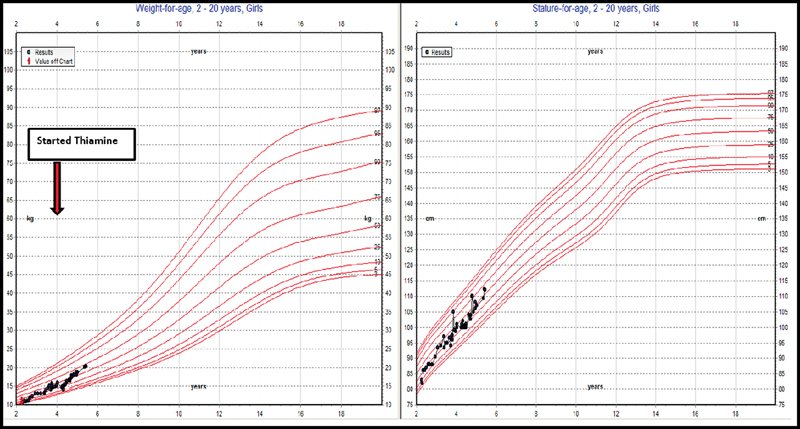
The patient's hemoglobin levels normalized to 12.9 g/dL without further blood transfusion, and insulin therapy was continued with reasonable control, with HbA1c of 6.4 to 7.7 g/dL. Subsequent follow-up showed a blood glucose range from 9.6 to 16.1 mmol/L and HbA1c from 6.4 to 7.7 mmol/L ([Table 1]). She then began to gain weight, as shown in the growth chart in [Fig. 3].
Her diagnosis was based on clinical findings, excluding other megaloblastic anemia causes and including the evidence of an excellent response to thiamine therapy.
Discussion
Our patient was diagnosed with classical TRMA, presenting with megaloblastic anemia, monogenic DM, sensorineural deafness, and thalassemia trait. This diagnosis was established on clinical background alone, as the parents refused genetic testing, and it was thus impossible to confirm the diagnosis. The 8-year-old patient has been treated with oral thiamine 100 mg twice daily, and her megaloblastic anemia improved. About 40 SLC19A2 mutations have been characterized, among which most are missense, nonsense, or insertions and deletions.[2] Such mutations have been described to lead to abnormal production of either aberrant or absent thiamine transporter protein, causing phenotype variations, and have been linked to TRMA presenting as a spectrum of symptoms and associated conditions. Other mutations may cause protein misfolding, interrupting its integration with the cell membrane. Regardless of the precise mechanisms, these mutations prevent the thiamine transporter protein from incorporating thiamine into the cell.[2] Monogenic DM is a rare DM type due to a single gene that causes early-onset damage to pancreatic β cells.[6]
Early intervention with high doses of thiamine supplementation as a preventative treatment may delay DM onset, especially if such treatment is started early after birth or even prenatally. Sensorineural deafness occurs progressively in early life and is irreversible; it is also unclear whether high thiamine supplement doses could improve or delay the hearing loss. There has been a single report of a patient who received thiamine doses during their first 2 months after birth and did not develop hearing loss.[5] Interestingly, animal model studies indicate that gradual hearing loss might be reversed following appropriate supplementation with thiamine as a prospective treatment plan.[6] In terms of clinical presentation, other than the typical triad of DM, megaloblastic anemia, and sensorineural deafness, other clinical features, such as a myriad of cardiovascular anomalies, optic and retinal atrophy, short stature, and hepatosplenomegaly, have also been described in TRMA cases. Retinitis pigmentosa was also reported in an African American female with TRMA.[7]
Thiamine, or vitamin B1, is a safe medication with no side effects or toxicity from possible overdosing. If started early in life and as soon as the diagnosis is suspected, response to thiamine treatment eliminates the need for repeated blood transfusions.[2] [8] Furthermore, high-dose thiamine therapy significantly treats anemia, achieves euglycemic control, and helps minimize or stop β-cell depletion. The hemoglobin level in one patient increased to 12.9 g/dL, but insulin therapy was still required.[2] [9] Even with a high thiamine therapy dose, hearing loss may be progressive, irreversible, or unresponsive, and it is worth noting that cochlear implantation may be beneficial for improving hearing loss, with significant benefits.[10]
Although it is simple, early thiamine therapy, when suspecting TRMA, may suffice to improve anemia and aid euglycemic control. Mixed blood film findings, including macrocytosis and microcytosis, should alert the clinician to suspect an association with hemoglobinopathies. Genetic analysis, including family screening, should be performed, and genetic counseling should be provided when possible.
Conclusion
To our knowledge, this is the first reported case of Rogers' syndrome associated with the thalassemia trait. A high index of suspicion of TRMA should always be considered in the differential diagnosis of macrocytic anemia comorbid with monogenic DM and sensorineural deafness. This case highlights the need for, and the importance of, early diagnosis and subsequent treatment with thiamine as soon as the diagnosis is suspected of reversing anemia and improving glycemic control potentially.
Conflict of Interest
None declared.
Acknowledgments
We acknowledge the help and support from the patients' parents.
Author Contributions
The authors contributed equally to the study.
Compliance with Ethical Principles
No prior ethical approval is required for single case reports.
Declaration of Patient Consent
Appropriate consent was obtained.
-
References
- 1 Porter FS, Rogers LE, Sidbury Jr JB. Thiamine-responsive megaloblastic anemia. J Pediatr 1969; 74 (04) 494-504
- 2 Spehar Uroic A, Milenkovic D, De Franco E, Bilic E, Rojnic Putarek N, Krnic N. Importance of immediate thiamine therapy in children with suspected thiamine-responsive megaloblastic anemia-report on two patients carrying a novel SLC19A2 gene mutation. J Pediatr Genet 2020; 11 (03) 236-239
- 3 Sanyoura M, Philipson LH, Naylor R. Monogenic diabetes in children and adolescents: recognition and treatment options. Curr Diab Rep 2018; 18 (08) 58
- 4 Amr K, Pawlikowska P, Aoufouchi S, Rosselli F, El-Kamah G. Whole exome sequencing identifies a new mutation in the SLC19A2 gene leading to thiamine-responsive megaloblastic anemia in an Egyptian family. Mol Genet Genomic Med 2019; 7 (07) e00777
- 5 Beshlawi I, Al Zadjali S, Bashir W, Elshinawy M, Alrawas A, Wali Y. Thiamine responsive megaloblastic anemia: the puzzling phenotype. Pediatr Blood Cancer 2014; 61 (03) 528-531
- 6 Shaw-Smith C, Flanagan SE, Patch A-M. et al. Recessive SLC19A2 mutations are a cause of neonatal diabetes mellitus in thiamine-responsive megaloblastic anaemia. Pediatr Diabetes 2012; 13 (04) 314-321
- 7 Lagarde WH, Underwood LE, Moats-Staats BM, Calikoglu AS. Novel mutation in the SLC19A2 gene in an African-American female with thiamine-responsive megaloblastic anemia syndrome. Am J Med Genet A 2004; 125A (03) 299-305
- 8 Manimaran P, Subramanian VS, Karthi S. et al. Novel nonsense mutation (p.Ile411Metfs*12) in the SLC19A2 gene causing thiamine responsive megaloblastic anemia in an Indian patient. Clin Chim Acta 2016; 452: 44-49
- 9 Alzahrani AS, Baitei E, Zou M, Shi Y. Thiamine transporter mutation: an example of monogenic diabetes mellitus. Eur J Endocrinol 2006; 155 (06) 787-792
- 10 Onal H, Bariş S, Ozdil M. et al. Thiamine-responsive megaloblastic anemia: early diagnosis may be effective in preventing deafness. Turk J Pediatr 2009; 51 (03) 301-304
Address for correspondence
Publication History
Article published online:
28 August 2023
© 2023. The Libyan Biotechnology Research Center. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
-
References
- 1 Porter FS, Rogers LE, Sidbury Jr JB. Thiamine-responsive megaloblastic anemia. J Pediatr 1969; 74 (04) 494-504
- 2 Spehar Uroic A, Milenkovic D, De Franco E, Bilic E, Rojnic Putarek N, Krnic N. Importance of immediate thiamine therapy in children with suspected thiamine-responsive megaloblastic anemia-report on two patients carrying a novel SLC19A2 gene mutation. J Pediatr Genet 2020; 11 (03) 236-239
- 3 Sanyoura M, Philipson LH, Naylor R. Monogenic diabetes in children and adolescents: recognition and treatment options. Curr Diab Rep 2018; 18 (08) 58
- 4 Amr K, Pawlikowska P, Aoufouchi S, Rosselli F, El-Kamah G. Whole exome sequencing identifies a new mutation in the SLC19A2 gene leading to thiamine-responsive megaloblastic anemia in an Egyptian family. Mol Genet Genomic Med 2019; 7 (07) e00777
- 5 Beshlawi I, Al Zadjali S, Bashir W, Elshinawy M, Alrawas A, Wali Y. Thiamine responsive megaloblastic anemia: the puzzling phenotype. Pediatr Blood Cancer 2014; 61 (03) 528-531
- 6 Shaw-Smith C, Flanagan SE, Patch A-M. et al. Recessive SLC19A2 mutations are a cause of neonatal diabetes mellitus in thiamine-responsive megaloblastic anaemia. Pediatr Diabetes 2012; 13 (04) 314-321
- 7 Lagarde WH, Underwood LE, Moats-Staats BM, Calikoglu AS. Novel mutation in the SLC19A2 gene in an African-American female with thiamine-responsive megaloblastic anemia syndrome. Am J Med Genet A 2004; 125A (03) 299-305
- 8 Manimaran P, Subramanian VS, Karthi S. et al. Novel nonsense mutation (p.Ile411Metfs*12) in the SLC19A2 gene causing thiamine responsive megaloblastic anemia in an Indian patient. Clin Chim Acta 2016; 452: 44-49
- 9 Alzahrani AS, Baitei E, Zou M, Shi Y. Thiamine transporter mutation: an example of monogenic diabetes mellitus. Eur J Endocrinol 2006; 155 (06) 787-792
- 10 Onal H, Bariş S, Ozdil M. et al. Thiamine-responsive megaloblastic anemia: early diagnosis may be effective in preventing deafness. Turk J Pediatr 2009; 51 (03) 301-304