

Subscribe to RSS
DOI: 10.1055/s-0043-1775398
Synthesis of 5-(4-Formylphenyl)barbituric Acid to Access Enolizable Chromophoric Barbituric Acids
Authors
Financial support from the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (DFG; SP 392/26-3) is gratefully acknowledged.

Dedicated to Prof. Dr. Stefan Spange
Abstract
Barbituric acids mono-substituted at the 5-position show keto–enol tautomerism. In the keto form, conjugation to an aryl substituent is interrupted due to the sp³-hybridized carbon atom at the 5-position of barbituric acid. The enol form generates a conjugated π-system to the aryl substituent and acts as an electron-donating group. If the aryl substituent is electron-deficient, a push-pull system is generated that shows typical UV/Vis absorption. These types of compounds are difficult to access synthetically due to their intrinsic convertibility. The synthesis of barbituric acids with a 4-formylphenyl functionality at the 5-position is reported. This compound, 5-(4-formylphenyl)barbituric acid, could be used to introduce extended π-systems with electron-withdrawing groups in great variety by simple condensation reactions. We demonstrate this by a Horner–Wadsworth–Emmons reaction that forms the enolizable dye (E)-5-(4-(4-nitrostyryl)phenyl)barbituric acid.
Barbituric acids with electron-withdrawing functionalities at the 5-position are suitable sensors to study the complex formation of receptor molecules with different hydrogen-bond patterns.[1] The complementary hydrogen-bond pattern of the receptor molecules changes the tautomerism of barbituric acid between the colorless keto form and the colored enol form.[1] The keto form has a hydrogen-bond pattern of a hydrogen-bond accepting group, a hydrogen-bond donating group, and a hydrogen-bond accepting group (ADA). In contrast, the enol form has a hydrogen-bonding pattern of a hydrogen-bond donating group, another hydrogen-bond donating group, and a hydrogen-bond accepting group (DDA) (compare Figure [1]).

The existing synthetic processes are only suitable for certain compounds. The described 5,4-nitrophenylbarbituric acid could only be synthesized by direct nitration of 5-phenylbarbituric acid,[1a] while 5-(2,4-dinitro)phenylbarbituric acid and 5-(4-((1,3-dioxo-1H-inden-(3H)-ylidene)methyl)phenyl)barbituric acid were accessible by nucleophilic aromatic substitution of the corresponding electrophilic compounds with barbiturate anions.[1b] The limitations of this method were demonstrated using 5-(4-((1,3-dioxo-1H-inden-(3H)-ylidene-(3H)-ylidene)methyl)phenyl)barbituric acid as an example. The side reaction at the methine carbon atom of the electrophile limits the synthesis. Only products for the alkylated barbiturate anions could be obtained because of their higher nucleophilicity.[2]
Therefore, a more flexible synthetic procedure is needed to reach a wider variety of functionalities. To introduce electron-withdrawing groups, condensation reactions of CH-acidic molecules with aldehydes are commonly used. The target molecule 5-(4-formylphenyl)barbituric acid would therefore allow the introduction of several different electron-withdrawing functionalities and different sizes of conjugated π-systems.
It should be noted that several approaches to synthesize 5-(4-formylphenyl)barbituric acid have failed. For example, direct Vilsmeier–Haack formylations[3] of 5-phenylbarbituric acid, the three-fold trimethylsilyl-protected 5-phenylbarbituric acid,[4] or 2-phenyldiethyl malonate were not successful. Pd-catalyzed C–C-coupling reactions[5] starting from 5-bromobarbituric acid[6] were also unsuccessful.
Thereby, a step-by-step synthetic procedure beginning from ethyl p-tolylacetate was established as shown in Figure [2]. The first step is the reaction of ethyl p-tolylacetate with diethyl carbonate under basic conditions.[7] Afterwards, the introduction of the formyl group begins with a Wohl–Ziegler bromination[8] followed by a Sommelet reaction,[9] which oxidizes the benzyl halide to an aldehyde. After protection of the aldehyde with ethylene glycol,[10] the diethyl malonate functionality could be condensed under basic conditions (sodium ethoxide) with urea to form the barbituric acid.[11] The resulting sodium barbiturate was filtered off, washed with ethanol, and dissolved in 1 M HCl to remove the protecting group and protonate the barbiturate. Commonly, barbituric acids are accessible by condensation reactions of diethyl malonate with urea.[11] [12] This procedure allows different functionalities to be introduced in the 5-position by substituted malonates[13] or N,N′-substitution starting with the respective alkylated urea.[14] This reaction was the final step in our synthetic route, which facilitates the synthesis of N,N′-alkylated barbituric acids as important compounds for the study of hydrogen bonding.[1]

Aldehyde 5 shows the envisioned keto–enol tautomerism in polar solvents such as DMSO, which is seen within the 1H NMR spectra (see the Supporting Information). The tautomerism lies with 40:60 slightly on the enol side (determined via integration of the respective signals at the 1H NMR spectrum). This observed keto–enol tautomerism as well as the high acidity of the barbituric acid derivates are the reasons for the broad signals in the obtained NMR spectra (see the Supporting Information). Enolizable barbituric acids with stronger electron-withdrawing groups, such as 5-(4-nitrophenyl)barbituric acid switched completely to the enol form.[1a] It could be shown that keto–enol tautomerism of barbituric acids depends on the substituent at the 5-position.[15] Additionally, the electron-withdrawing effect of the substituents influences the keto–enol tautomerism strongly. An appropriate strength of the electron-withdrawing group of the functionality is important because the tautomerism must be easily changeable by complementary hydrogen-bonding sequences to either the keto- or enol-form of the barbituric acid to be a suitable sensor molecule (Figure [1]).
Aldehyde 5 could be used for condensation reactions to increase the conjugated π-system and to introduce electron-withdrawing groups. For example, we demonstrate this procedure by a Horner–Wadsworth–Emmons reaction with diethyl 4-nitrobenzylphosphonate to form (E)-5-(4-(4-nitrostyryl)phenyl) barbituric acid (6) (Figure [3]).

Compound 6 shows the envisioned keto–enol tautomerism during NMR experiments in DMSO. The stereochemistry of the C–C double bond of the neutral form could not be determined via 1H NMR experiments because of the observed keto–enol tautomerism of the compound. However, by deprotonation of compound 6 to generate the barbiturate anion with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), a coupling constant of 16 Hz for the C–C double bond could be found, which indicates the trans-conformation of the stilbene (see the Supporting Information).
The acidity constant (pK a) of compound 6 was determined by pH-dependent UV/Vis titration in water/methanol (5:1) at 20 °C, conducted with a pH-immersion probe. The measurements commenced with a 0.5 M sodium hydroxide solution (see the Supporting Information). The pK a values of compound 6 were calculated using the Henderson–Hasselbalch equation (Equation 1).[16]

The absorbance of 6, as well as the absorbance of the corresponding anion, were set as the respective concentrations c HA and c A⊝ (see the Supporting Information). Additionally, a second deprotonation was found, which is typical for deprotonation of NH-functionalities. Compound 6 deprotonates at pH 1.90 and forms a dianion at pH >12. Table [1] shows the pK a values of 6 compared with reported data for enolizable barbituric acids.
 |
||||
|
pK a (1) |
pK a (2) |
Lit. |
||
|
R1 = R2 = H (6) |
 |
1.90 |
12.80 |
this paper |
|
R1 = R2 = H |
 |
2.54 |
–c |
[17] |
|
R1 = R2 = H |
 |
1.80 |
12.06 |
[18] |
|
R1 = n-Bu; R2 = H |
1.93 |
12.94 |
[18] |
|
|
R1 = R2 = Me |
2.65 |
–d |
[18] |
|
|
R1 = n-Bu; R2 = H |
 |
1.35 |
–b |
[1b] |
|
R1 = R2 = Me |
1.34 |
–b |
[1b] |
|
|
R1 = R2 = H |
 |
0.14 |
11.76 |
[1b] |
|
R1 = n-Bu; R2 = H |
0.13 |
12.42 |
[1b] |
|
|
R1 = R2 = Me |
–0.23 |
–d |
[1b] |
|
|
R1 = R2 = H |
 |
–2.60 |
–c |
[19] |
a pK a value (1) arises from the acidic CH-group and pK a (2) from the NH functionality.
b Decolorization at pH >6, caused by a reversible nucleophilic attack at the methine carbon.[1b]
c Not reported in the literature.
d Dialkylated barbituric acid – no second acidic proton.
Table [1] shows clearly that the acidity increases with stronger electron-withdrawing groups as well as smaller π-systems separating the electron-withdrawing group from the acidic proton. Thereby, the acidity decreases from 5-nitrobarbituric acid (–2.60) to 5-(4-nitrophenyl)barbituric acid (1.80) and slightly to (E)-5-(4-(4-nitrostyryl)phenyl) barbituric acid 6 (1.90).
Solvatochromic investigations of compound 6 were carried out in 21 different solvents to describe the influence of different interactions on the chromophore. The linear solvation energy relationships (LSER) of Kamlet and Taft[20] (Equation 2) and Catalán[21] [22] [23] [24] (Equation 3) were used to evaluate the solvatochromic studies.

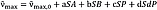
In the case of enolizable dyes, or otherwise variable chromophores, it is important to point out that solvatochromism could always just describe one chromophoric system. Therefore, special care must be taken to ensure that a UV/Vis absorption band can always be assigned to the same chromophoric system. Mixing UV/Vis absorptions of different species, for example, the keto and enol forms of compound 6, leads to incorrect results of the multiple square correlation analysis.
The UV/Vis absorptions of the enol species of 6 were found in the region 340–380 nm. Because of the missing electron-donating effect of the enol-group, the keto-species is hypsochromically shifted and was found at wavelengths of 300 nm and less. It is also possible that solvents deprotonate the barbituric acid. The generated barbiturate anion is a much stronger electron-donating group and was detected by adding DBU to a solution of 6 in DMSO, for example at 474 nm. The UV/Vis absorption maxima of the enolic species of 6 in different solvents were analyzed and are summarized in the Supporting Information.
Multiple linear correlation analysis results in Equation 4 and Equation 5. The negative correlation coefficients indicate that a bathochromic shift appears if the solvent is hydrogen-bond accepting or polar. The Catalán equation also indicates a bathochromic shift for acidic / hydrogen-bond donating solvents. This effect could not be found in the Kamlet–Taft equation (a ≅ 0).


The bathochromic shift found for hydrogen-bond-accepting solvents could be interpreted as interaction with the NH-functionality of the barbituric acid or the enolic OH. Both interactions would increase the electron density at the electron donating side of the chromophore’s push-pull system and result in the observed bathochromic shift of the UV/Vis absorption. This is a clear indication that the enolic form of the compound has been investigated. The bathochromic shift caused by acidic solvents found within the Catalán equation (Equation 5) is mainly caused by a hydrogen bond towards the nitro-group, which increases the electron-withdrawing effect. Interactions with the carbonyl oxygens of the barbituric acid, especially with the enol-function, would result in a hypsochromic shift. Those interactions are superimposed by the bathochromic shift caused by the nitro group. The graphically depicted results of the multiple square correlation analyses are shown in Figure [4] (Kamlet–Taft) and Figure [5] (Catalán).


Most recently, Spange and co-workers found that HBD-parameters also represent volumetric effects of the collective hydrogen bonds on the probe, as well as the pK a of the solvent and had to be included in the considerations.[25] These effects could also explain the results found in the correlations by Kamlet–Taft and Cátalan (Equations 2 and 3). Especially, for acidic dyes such as the enolizable barbituric acids, the pK a of the solvent can play the main role in describing the observed solvatochromic shifts,[1b] which is why acid-base reactions must be consistently excluded.
Summarized, it could be said that the solvatochromism of compound 6 is dominated by nonspecific interactions. The coefficients for solvent polarizability and dipolarity are negative, indicating a more polar first exited state, which is stabilized by polar solvents. Specific interactions with hydrogen-bond-accepting or -donating solvents are of less importance, as indicated by the small coefficients. This means that the formation of hydrogen bonds does not influence the UV/Vis absorption of the enolic barbiturate 6 significantly. This result is expected, because the larger conjugated π-system usually results in a smaller solvatochromic range of the dye.[26]
For the usability of our chromophore barbituric acid 6 as a sensor molecule for nucleobases, this result shows the importance of the keto–enol tautomerism caused by the complementary hydrogen-bonding pattern. The effect of solvatochromism on the UV/Vis absorption maximum (Δν̃max) of compound 6 is only 1750 cm–1 (348–370 nm). The shift caused by the keto–enol tautomerism on the other hand is large. In several solvents such as methanol, the absorption of the keto form could be detected. In the example of methanol, the keto form was found at 271 nm while the enol form was bathochromically shifted to 353 nm. This shift of 8570 cm–1 (82 nm) is seen in a change from colorless to yellow. These optical properties combined with the comparable low acidity (pK a 1.90) of an enolizable barbituric acid, make compound 6 a suitable sensor dye for nucleobases and shows the importance of increasing the conjugated π-system of enolizable barbituric dyes.
Based on aldehyde 5, further enolizable barbituric acid dyes can be synthesized. The nitrostilbene 6 as well as the indene[1b] already show that chromophores with an increased conjugated π-system are better suited as sensor molecules for nucleobases because of their lower acidity. It is important to point out that the smaller solvatochromic range of these dyes does not restrict their use as chromophoric sensors because the switch between keto and enol form still results in a strong shift of the UV/Vis absorption.
Solvents and reagents were purchased from Aldrich, Riedel-de Haёn, Merck, Acros, Roth, Fluka, and ABCR. Reagents were used without further purification. Solvents were dried with standard procedures (dichloromethane and ethanol) or purchased as extra dry (DMF). Solvents for UV/Vis measurements were dried with standard procedures and stored under an argon atmosphere or purchased as spectroscopic grade. Solvent mixtures are indicated as volume mixtures (v/v).
NMR spectra were recorded with a Bruker Avance 250 spectrometer and reported in ppm relative to TMS with internal reference to the respective solvent (CDCl3: 1H δ = 7.26 ppm, 13C δ = 77.16 ppm; d 6-DMSO: 1H δ = 2.50 ppm, 13C δ = 39.52 ppm). Elemental analyses were carried out with a Vario EL of Elementar Analysegeräte GmbH. UV/Vis spectra were recorded with a MCS 400 spectrometer from Carl Zeiss Jena GmbH with a spectral range of 210–1010 nm in 1 cm quartz glass cuvettes.
2-Tolyldiethyl Malonate (1)
To a solution of diethyl carbonate (65 mL, 0.5 mol) in anhydrous THF (200 mL), sodium hydride (6.74 g, 0.281 mol) was added in small portions. A solution of ethyl p-tolylacetate (20.0 g, 0.112 mol) in anhydrous THF (50 mL) was then slowly added dropwise. The solution was stirred for 2 h under reflux. After cooling, the THF solution was washed with ammonium chloride (2 × 100 mL) and the aqueous layer was extracted with diethyl ether (100 mL). The combined organic layers were washed with NaHCO3 (2 × 50 mL), brine, and finally with water. The organic layer was dried over MgSO4 and the solvent was removed under reduced pressure. The obtained crude product was distilled (0.2 mbar at 108 °C) to give 2-tolyldiethyl malonate (1).
Yield: 19.36 g (69%); colorless oil.
NMR spectra data were identical to those reported in the literature.[27]
1H NMR (250 MHz, CDCl3): δ = 1.27 (t, 3 J HH = 7.25 Hz, 6 H), 2.35 (s, 3 H), 4.22 (m, 4 H), 4.58 (s, 1 H), 7.18 (d, 3 J HH = 8.0 Hz, 2 H), 7.30 (d, 3 J HH = 8.0 Hz, 2 H).
13C NMR (62.9 MHz, CDCl3): δ = 13.5 (CH3), 20.7 (CH3), 57.1 (CH), 61.3 (CH2), 128.6 (CH), 128.8 (CH), 129.4 (C), 137.5 (C), 167.9 (C).
Anal. Calcd for C14H18O4: C, 67.18; H, 7.25. Found: C, 67.67; H, 7.30.
2-(4-Bromomethylphenyl)diethyl Malonate (2)
To a solution of 2-(4-tolyl)diethyl malonate (4.94 g, 19.74 mmol) in CCl4 (30 mL), N-bromosuccinimide (3.74 g, 21.0 mmol) was added under stirring. Azobisisobutyronitrile (50 mg, 0.30 mmol) was then added and the solution was stirred for 7 h under reflux. After cooling, the precipitating side product succinimide was filtered off. The solvent was removed under reduced pressure to yield product 2 as yellowish oil (5.39 g, 83%). The crude product was used in the following steps without further purification. NMR spectra data were identical to those reported in the literature.[8]
1H NMR (250 MHz, CDCl3): δ = 1.26 (t, 3 J HH = 7.5 Hz, 6 H), 4.21 (m, 4 H), 4.48 (s, 2 H), 4.60 (s, 1 H), 7.39 (s, 4 H).
13C NMR (62.9 MHz, CDCl3): δ = 13.5 (CH3), 32.5 (CH2Br), 57.1 (CH), 61.5 (CH2), 128.8 (CH), 129.3 (CH), 132.5 (C), 137.2 (C), 167.4 (C).
2-(4-Formylphenyl)diethyl Malonate (3)
2-(4-Bromomethylphenyl)diethyl malonate (3.96 g, 12.0 mmol) was dissolved in 50% acetic acid (50 mL). Hexamethylenetetramine (1.08 g, 7.7 mmol) was added and the solution was stirred for 4 h under reflux. The solution was cooled and extracted with CH2Cl2 (3 × 30 mL). The combined organic phases were washed twice with water and dried over MgSO4. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (Kieselgel 60; n-pentane/EtOAc, 22:3). During the bromination, unreacted 2-(4-tolyl)diethyl malonate (1) could be isolated (Rf = 0.49) as well as 3 as a colorless oil (1.33 g, 42%, Rf = 0.37). NMR spectra data were identical to those reported in the literature.[28]
1H NMR (250 MHz, CDCl3): δ = 1.27 (t, 3 J HH = 7.2 Hz, 6 H), 4.23 (m, 4 H), 4.69 (s, 1 H), 7.59 (d, 3 J HH = 8.3 Hz, 2 H), 7.89 (d, 3 J HH = 8.3 Hz, 2 H), 10.02 (s, 1 H).
13C NMR (62.9 MHz, CDCl3): δ = 14.1 (CH3), 58.1 (CH2), 62.3 (CH2), 130.0 (CH), 130.2 (CH), 136.2 (C), 139.3 (C), 167.5 (C), 191.9 (CH).
Anal. Calcd for C14H16O5: C, 63.62; H, 6.11. Found: C, 62.35; H, 6.02.
2-(4-(1,3-Dioxolan-2-yl)phenyl)diethyl malonate (4)
2-(4-Formylphenyl)diethyl malonate (1.33 g, 5.05 mmol) was dissolved in CH2Cl2 (30 mL) and ethylene glycol (1.0 mL, 17.89 mmol) was added. Under stirring, p-toluenesulfonic acid (50 mg, 0.25 mmol) was added and the mixture was stirred for 5 h under reflux. The solution was cooled and washed three times with water. The organic phase was dried over MgSO4 and the solvent was removed under reduced pressure to give 4 (1.55 g, quant.) as a yellowish oil. The crude product was used in the next step without further purification.
1H NMR (250 MHz, CDCl3): δ = 1.27 (t, 3 J HH = 7.1 Hz, 6 H), 4.08 (m, 4 H), 4.20 (m, 4 H), 4.62 (s, 1 H), 5.82 (s, 1 H), 7.40–7.55 (m, 4 H).
5-(4-Formylphenyl)barbituric Acid (5)
Pieces of sodium (350 mg, 12 mmol) were added to an evaporated three-neck flask and absolute EtOH (20 mL) was added. The mixture was stirred until the sodium was reacted completely. Urea (1.00 g, 6.00 mmol) and 2-(4-(1,3-dioxolan-2-yl)phenyl)diethyl malonate (1.56 g, 5.05 mmol) were then added and the mixture was stirred for 7 h under reflux. During this time, the reaction turned intense yellow and a solid precipitate was seen. After cooling, the solid was filtered off and washed with cold ethanol. To remove the protecting group, the solid was dissolved in 1 M HCl and cooled. After 30 min, the precipitate was filtered off, washed intensively with water and dried under vacuum to give 5 (199 mg, 17%) as a yellowish solid.
1H NMR (250 MHz, DMSO-d 6): δ (keto/enol) = 5.15 (s, 0.5 H, CH-keto), 7.50–7.90 (m, 4 H), 9.94 (s, 1 H), 10.58 (s, 1.1 H), 11.46 (s, 0.8 H).
13C NMR (62.9 MHz, DMSO-d 6): δ (keto/enol) = 55.1 (C(5)H-keto), 91.4 (C(5)-enol), 124.1, 128.6, 129.5, 130.7, 131.0, 133.4, 139.9, 140.6 (C-Ar), 149.8 (CO), 150.9 (CO), 161.6 (CO), 168.6 (CO), 192.6 (CH), 192.7 (CH).
Anal. Calcd for C11H8O4N2: C, 56.90; H, 3.47; N, 12.07. Found: C, 56.63; H, 3.80; N, 11.89.
(E)-5-(4-(4-Nitrosyryl)phenyl)barbituric Acid (6)
Under inert conditions and cooling to 0 °C, diethyl-4-nitrobenzyl phosphonate (118 mg, 0.431 mmol) and potassium tert-butoxide (55 mg, 0.474 mmol) were dissolved in anhydrous DMF (3 mL) (the solution became red, indicating the formation of the carbanions). After 15 min stirring, 5-(4-formylphenyl)barbituric acid (100 mg, 0.431 mmol) dissolved in anhydrous DMF (2 mL) was added. The mixture was stirred for 16 h at r.t. then dropped in ice-cold 1 M HCl. The precipitated yellow solid was filtered off and washed intensively with water and dried under reduced pressure to give 6 (50 mg, 33%) as a yellow solid.
1H NMR (250 MHz, DMSO-d 6): δ (keto/enol) = 4.90 (s, 0.4 H, CH-keto), 7.30–7.80 (m, 6 H), 7.84 (d, 3 J HH = 8.5 Hz, 2 H), 8.21 (d, 3 J HH = 8.5 Hz, 2 H), 9.76 (s, 0.7 H), 11.42 (s, 0.7 H).
Because of the keto-enol equilibrium of 6, characterization by NMR was difficult. Therefore, the sodium salt 6a was generated in situ and characterized by NMR analysis. The coupling constant of 16 Hz, clearly shows the trans-configuration of the stilbene.
1H NMR (250 MHz, DMSO-d 6): δ (anion) = 7.21 (d, 3 J HH = 16 Hz, 1 H), 7.40 (d, 3 J HH = 8.5 Hz, 2 H), 7.44 (d, 3 J HH = 16 Hz, 1 H), 7.81 (d, 3 J HH = 9 Hz, 2 H), 8.07 (d, 3 J HH = 8.5 Hz, 2 H), 8.19 (d, 3 J HH = 9 Hz, 2 H), 9.18 (s, 2 H).
13C NMR (62.9 MHz, DMSO-d 6): δ (anion) = 86.8 (C), 122.5 (CH), 124.1 (CH), 125.4 (CH), 126.6 (CH), 128.6 (CH), 129.0 (C), 134.6 (CH), 140.8 (C), 145.1 (C), 145.4 (C), 151.2 (C), 164.1 (C).
Conflict of Interest
The author declares no conflict of interest.
Acknowledgment
The author acknowledges the support of Prof. Dr. S. Spange for his support and discussion. He always describes the 5-(4-formylphenyl)barbituric acid as the Mt. Everest of barbituric acids. Further acknowledgments for fruitful discussions and support to Dr. M. Bauer, Dr. K. Schreiter, and Dr. A. Oehlke.
Supporting Information
- Supporting information for this article is available online at https://doi.org/10.1055/s-0043-1775398.
- Supporting Information (PDF) (opens in new window)
-
References
- 1a Bolz I, Spange S. Chem. Eur. J. 2008; 14: 9338
- 1b Schade A, Schreiter K, Rüffer T, Lang H, Spange S. Chem. Eur. J. 2016; 22: 5734
- 2 Schade A, Tchernook I, Bauer M, Oehlke A, Breugst M, Friedrich J, Spange S. J. Org. Chem. 2017; 82: 8476
- 3a Vilsmeier A, Haack A. Ber. Dtsch. Chem. Ges. 1927; 60: 119
- 3b Marson CM. Tetrahedron 1992; 48: 3659
- 3c Reichardt C. J. Prakt. Chem. 1999; 341: 609
- 4 Studentsov EP, Kokhanovskii AN, Ganina MB, Nikolaeva NI, Fedorova EV, Moskvin AV, Ivin BA. Russ. J. Gen. Chem. 2004; 74: 261
- 5 Miyaura N, Suzuki A. Chem. Rev. 1995; 95: 2457
- 6 Bright R, Coote SJ, Freeman S, Hayes D, Smith G, Tapolczay D. Synth. Commun. 1996; 26: 4195
- 7 Chênevert R, Dasjardins M. Can. J. Chem. 1994; 72: 2312
- 8 Sang-Uk K, Worthy KM, Bindu LK, Zhang M, Yang D, Fisher RJ, Burke TR. Jr. J. Med. Chem. 2005; 48: 5369
- 9 Salama TA, Lackner B, Falk H. Monatsh. Chem. 2003; 134: 1113
- 10 Meskens AJ. Synthesis 1981; 7: 501
- 11 Dickey JB, Gray AR. Org. Synth. 1938; 18: 8
- 12 Baeyer A. Justus Liebigs Ann. Chem. 1862; 127: 1
- 13 Mahmudov KT, Kopylovich MN, Maharramov AM, Kurbanova MM, Gurbanov AV, Pombeiro AJ. L. Coord. Chem. Rev. 2014; 265: 1
- 14 Stein A, Gregor HP, Spoerri PE. J. Am. Chem. Soc. 1956; 78: 6185
- 15 Koffer H. J. Chem. Soc., Perkin Trans. 2 1975; 819
- 16a Henderson LJ. Am. J. Physiol. 1908; 21: 173
- 16b Hasselbalch KA. Biochem. Zeitschrift 1918; 78: 112
- 17 Koffer H. J. Chem. Soc., Perkin Trans. 2 1974; 1428
- 18 Bolz I. Dissertation . Technische Universität Chemnitz; Chemnitz: 2009. https://nbn-resolving.org/urn:nbn:de:bsz:ch1-200901505
- 19 Slesarev VI, Popov AS. Russ. J. Gen. Chem. 2000; 70: 615
- 20 Kamlet MJ, Abboud J.-LM, Taft RW. J. Am. Chem. Soc. 1977; 99: 6027
- 21a Catalán J, Díaz C. Liebigs Ann./Recl. 1997; 1941
- 21b Catalán J, Díaz C. Eur. J. Org. Chem. 1999; 885
- 22a Catalán J, Díaz C, López V, Pérez P, de Paz J.-LG, Rodríguez JG. Liebigs Ann. 1996; 1785
- 22b Catalán J, Palomar J, Díaz C, de Paz J.-LG. J. Phys. Chem. A 1997; 101: 5183
- 23 Catalán J, Hopf H. Eur. J. Org. Chem. 2004; 4694
- 24 Catalán J. J. Phys. Chem. B 2009; 113: 5951
- 25 Spange S, Weiß N. ChemPhysChem 2023; 24: e202200780
- 26 Reichardt C. In Solvents and Solvent Effects in Organic Chemistry, Vol. 3. Wiley-VCH; Weinheim: 2003: 329
- 27 Guanti G, Narisano E, Podgorski T, Thea S, Williams A. Tetrahedron 1990; 46: 7081
- 28 Melngaile R, Videja M, Kuka J, Kinens A, Zacs D, Veliks J. Org. Lett. 2023; 25: 2280
Corresponding Author
Publication History
Received: 05 June 2024
Accepted after revision: 03 July 2024
Article published online:
02 September 2024
© 2024. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1a Bolz I, Spange S. Chem. Eur. J. 2008; 14: 9338
- 1b Schade A, Schreiter K, Rüffer T, Lang H, Spange S. Chem. Eur. J. 2016; 22: 5734
- 2 Schade A, Tchernook I, Bauer M, Oehlke A, Breugst M, Friedrich J, Spange S. J. Org. Chem. 2017; 82: 8476
- 3a Vilsmeier A, Haack A. Ber. Dtsch. Chem. Ges. 1927; 60: 119
- 3b Marson CM. Tetrahedron 1992; 48: 3659
- 3c Reichardt C. J. Prakt. Chem. 1999; 341: 609
- 4 Studentsov EP, Kokhanovskii AN, Ganina MB, Nikolaeva NI, Fedorova EV, Moskvin AV, Ivin BA. Russ. J. Gen. Chem. 2004; 74: 261
- 5 Miyaura N, Suzuki A. Chem. Rev. 1995; 95: 2457
- 6 Bright R, Coote SJ, Freeman S, Hayes D, Smith G, Tapolczay D. Synth. Commun. 1996; 26: 4195
- 7 Chênevert R, Dasjardins M. Can. J. Chem. 1994; 72: 2312
- 8 Sang-Uk K, Worthy KM, Bindu LK, Zhang M, Yang D, Fisher RJ, Burke TR. Jr. J. Med. Chem. 2005; 48: 5369
- 9 Salama TA, Lackner B, Falk H. Monatsh. Chem. 2003; 134: 1113
- 10 Meskens AJ. Synthesis 1981; 7: 501
- 11 Dickey JB, Gray AR. Org. Synth. 1938; 18: 8
- 12 Baeyer A. Justus Liebigs Ann. Chem. 1862; 127: 1
- 13 Mahmudov KT, Kopylovich MN, Maharramov AM, Kurbanova MM, Gurbanov AV, Pombeiro AJ. L. Coord. Chem. Rev. 2014; 265: 1
- 14 Stein A, Gregor HP, Spoerri PE. J. Am. Chem. Soc. 1956; 78: 6185
- 15 Koffer H. J. Chem. Soc., Perkin Trans. 2 1975; 819
- 16a Henderson LJ. Am. J. Physiol. 1908; 21: 173
- 16b Hasselbalch KA. Biochem. Zeitschrift 1918; 78: 112
- 17 Koffer H. J. Chem. Soc., Perkin Trans. 2 1974; 1428
- 18 Bolz I. Dissertation . Technische Universität Chemnitz; Chemnitz: 2009. https://nbn-resolving.org/urn:nbn:de:bsz:ch1-200901505
- 19 Slesarev VI, Popov AS. Russ. J. Gen. Chem. 2000; 70: 615
- 20 Kamlet MJ, Abboud J.-LM, Taft RW. J. Am. Chem. Soc. 1977; 99: 6027
- 21a Catalán J, Díaz C. Liebigs Ann./Recl. 1997; 1941
- 21b Catalán J, Díaz C. Eur. J. Org. Chem. 1999; 885
- 22a Catalán J, Díaz C, López V, Pérez P, de Paz J.-LG, Rodríguez JG. Liebigs Ann. 1996; 1785
- 22b Catalán J, Palomar J, Díaz C, de Paz J.-LG. J. Phys. Chem. A 1997; 101: 5183
- 23 Catalán J, Hopf H. Eur. J. Org. Chem. 2004; 4694
- 24 Catalán J. J. Phys. Chem. B 2009; 113: 5951
- 25 Spange S, Weiß N. ChemPhysChem 2023; 24: e202200780
- 26 Reichardt C. In Solvents and Solvent Effects in Organic Chemistry, Vol. 3. Wiley-VCH; Weinheim: 2003: 329
- 27 Guanti G, Narisano E, Podgorski T, Thea S, Williams A. Tetrahedron 1990; 46: 7081
- 28 Melngaile R, Videja M, Kuka J, Kinens A, Zacs D, Veliks J. Org. Lett. 2023; 25: 2280