

Subscribe to RSS
DOI: 10.1055/s-0044-1780495
Efficient and Scalable Enantioselective Synthesis of a Key Intermediate for Rimegepant: An Oral CGRP Receptor Antagonist
Authors
Funding The work was supported by Enterprise Key Laboratory of Anti-viral Drug Development for Guangdong Province (Grant No. 2020B1212070003).


Abstract
Rimegepant is a calcitonin gene-related peptide antagonist used for acute treatment and prevention of migraine. We herein attempt to explore an efficient and practiced method for scale-up, regio- and enantioselective synthesis of (R)-9-hydroxy-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (1), a key intermediate of rimegepant. In this work, a Ru-catalyzed asymmetric transfer hydrogenation (ATH) reaction was a key step. The optimization of the reaction conditions involved exploring the reaction parameters including catalysts, bases, and solvents. The results suggested that the Ru-catalyzed ATH process using formic acid as the hydrogen donor could be operated under mild conditions at a low catalyst loading (0.5 mol%), affording a high yield (92.1% yield with 99.8% purity) and gratifying enantioselectivity (99.9% ee) of the target product (1). This work first reported the Ru-catalyzed ATH process in the synthesis of key intermediates of rimegepant. The optimized ATH process was easy to implement and cost-effective, making it particularly suitable for manufacturing scale production.
Keywords
rimegepant - enantioselective synthesis - Ru-catalyzed - asymmetric transfer hydrogenation reactionIntroduction
Rimegepant is a calcitonin gene-related peptide antagonist developed and launched by Biohaven Pharmaceutical (a wholly owned subsidiary of Pfizer) under license from Bristol-Myers Squibb. It is indicated for acute migraine treatment and prevention and has the potential to treat trigeminal neuralgia and chronic rhinosinusitis, for which studies are still ongoing.[1] [2] [3] [4] A cyclohepta[b]pyridine system of rimegepant has three chiral centers, as shown in [Fig. 1], [5] [6] [7] [8] and how to control the region and stereochemistry of the drug during its synthesis is extremely challenging.

(R)-9-Hydroxy-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (1), as a key intermediate of rimegepant,[7] [8] [9] is synthesized from 7,8-dihydro-5H-cyclohepta[b]pyridine-5,9(6H)-dione (2) by enantioselective ketone reduction ([Fig. 1]). Compound 2 has two carbonyl functional groups on the cyclohepta[b]pyridine ring, and the chemical environments of the two carbonyl groups are similar, except that the carbonyl group at position 9 is closer to the nitrogen atom of the pyridine ring, thus, choosing to selectively reduce only one of the ketone groups and at the same time achieving a high degree of chiral purity is extremely challenging.
The reported synthetic methods of compound 1 are listed in [Scheme 1]. Luo et al constructed the cyclohepta[b]pyridine skeleton (2) from the starting material pyridine-2,3-dicarboxylic acid (3), followed by the stereoselective enzyme reduction of the diketone intermediate to achieve compound 1 and its enantiomer (4), which had to convert the configuration through a Mitsunobu reaction to give the target product.[10] The tedious operation undoubtedly challenged the overall efficiency of the process. Leahy et al obtained the target product (1) via asymmetric hydrogenation of compound 2 applying ES-KRED-119 and Rh ((R)-binapine)-(COD)BF4 as catalysts, respectively.[5] When ES-KRED was used as a reductant, impurities 5 and 6 were generated in parallel, and the target product (1) was isolated at 99.2% ee in only 81% yield, interestingly, when Rh((R)-binapine)(COD)BF4 was used as a catalyst, the chemo- and enantioselectivity was significantly improved with 99.9% ee and 100% reaction conversion being obtained, but it is well known that the cost of Rh((R)-binapine)(COD)BF4 is very high due to the extreme rarity of rhodium. Guo et al performed Rh((R)-binapine)(COD)BF4-catalyzed asymmetric hydrogenation under 40 to 50 psi, which resulted in low enantioselectivity (only 90% ee, 97% yield), and they had to use camphorsulfonic acid as the resolution reagent and add one more chiral resolution step to improve the chiral purity of the product.[9] [11] [12] [13] Given the above, there is an urgent need to explore an efficient and cost-saving method for the enantioselective synthesis of the key intermediate 1.

The asymmetric transfer hydrogenation (ATH) reaction is a potential solution to meet the regio- and stereoselectivity challenge in the synthesis of compound 1 since the reactivity of two carbonyl functional groups adjacent to pyridine would be different under special catalytic reaction conditions. In this study, the process optimization of the reaction,[14] [15] [16] [17] [18] [19] [20] [21] including the screening of catalysts, solvents, bases, and hydrogen sources, was performed. When CAT05 was used as a catalyst, dichloromethane as a solvent, diisopropylethylamine as a base, and formic acid (HCOOH) as a hydrogen source, the process gave compound 1 with an overall yield of 92.1%, a high purity of more than 99.8%, and an ee value of 99.9%. This robust process can be operated under mild conditions, and the catalyst was replaced by a low-loading ruthenium catalyst (0.5% equiv.), which is much more affordable and readily available, resulting in significant cost advantages.
Results and Discussion
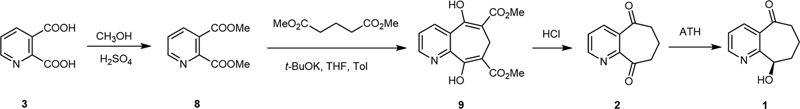
The designed synthesis route of compound 1 in this work is shown in [Scheme 2]. The commercially available compound 3 was esterified to give compound 8 followed by an ester condensation reaction to generate the cyclohepta[b]pyridine core (9) according to a reported study.[11] Subsequently, the decarboxylation reaction was performed in HCl (aq) providing compound 2, which was subjected to an ATH reaction to give the target compound (1).
The two carbonyl groups of compound 2 have a similar chemical environment, thus it is not easy to achieve a single pure configuration of the target product. The possible by-product is shown in [Scheme 3]. Then, we attempted to optimize ATH reaction conditions by screening the catalyst, bases, solvent, and hydrogen donors of the reaction.

As shown in [Scheme 4], when CAT01 (5% equiv.) was used as a catalyst, the starting material was consumed completely; however, liquid chromatography-tandem mass spectrometry showed no desired compound. The main product was diol side products. We then tried to decrease the amount of catalyst, the reaction temperature, and the amount of HCOOH. Fortunately, at room temperature, with CAT01 (0.2% equiv.) and HCOOH (1.09 equiv.), the stereoselectivity of the reaction was improved, obtaining 1 in 93.5% yield and 99.78% ee.

Considering the outsourcing catalyst CAT01 is very expensive, we are dedicated to seeking alternative catalysts that can be synthesized in-house. Therefore, different ligands for ruthenium catalysts were screened. As shown in [Table 1]. Among the catalysts (CAT02, CAT03, CAT04, and CAT05), CAT05 has the highest selectivity (99.44% ee) and conversion rate (93.53% yield) at a lower cost.
|
|
||||
|---|---|---|---|---|
|
Entry |
Catalyst |
Amount (%) |
Yield (%) |
ee (%)[b] |
|
1 |
CAT02 |
1 |
NA |
4.24 |
|
2 |
CAT03 |
1 |
75.37 |
98.32 |
|
3 |
CAT04 |
1 |
71.97 |
97.74 |
|
4 |
CAT05 |
0.5 |
93.53 |
99.44 |

a Compound 2, CT, and DCM were poured into the reaction vessel and purged with N2. Then, TEA and HCOOH were added. The mixture was stirred at room temperature for 18 hours. The crude product was purified by silica gel chromatography (n-hexane:EA = 4:1, 3:1), and concentrated under vacuum to dryness to give a product.
b It was detected by high-performance liquid chromatography using an IC column.
Different bases including TEA, diethylamine, diisopropylamine, and N,N-diisopropylethylamine (DIPEA) were screened. As shown in [Table 2], among the bases, DIPEA gave the highest enantioselectivity.
|
|
|||
|---|---|---|---|
|
Entry |
Base |
Yield (%) |
ee (%)[b] |
|
1 |
TEA |
93.53 |
99.44 |
|
2 |
Diethylamine |
/ |
93.28 |
|
3 |
Diisopropylamine |
/ |
99.26 |
|
4 |
DIPEA |
89.2 |
99.52 |

Abbreviations: DIPEA, N,N-diisopropylethylamine; TEA, triethylamine.
a Catalyst CAT05 (0.5%) and HCOOH (1.1 equiv.) were charged with the reaction vessel, and the reaction mixture was stirred at room temperature for 28.5 hours.
b It was detected by high-performance liquid chromatography employing an IC column.
Common solvents used in organic reactions including ethyl acetate, toluene, methanol, dichloromethane, ethyl alcohol, isopropyl alcohol, 1,2-dichloroethane, acetone, acetonitrile, and tetrahydrofuran were screened for the ATH reaction. As shown in [Table 3], when dichloromethane was used as a solvent, the reaction achieved the best performance with the highest yield (95.36%) and enantioselectivity (99.40%) obtained.
|
|
|||
|---|---|---|---|
|
Entry |
Solvent |
Yield (%) |
ee (%)[b] |
|
1 |
Ethyl acetate |
78.6 |
98.86 |
|
2 |
Toluene |
90.36 |
98.96 |
|
3 |
Methanol |
50.32 |
99.28 |
|
4 |
Dichloromethane |
95.36 |
99.40 |
|
5 |
Ethyl alcohol |
88.85 |
99.42 |
|
6 |
Isopropyl alcohol |
89.03 |
99.32 |
|
7 |
1,2-Dichloroethane |
91.62 |
99.48 |
|
8 |
Acetone |
63.44 |
98.54 |
|
9 |
Acetonitrile |
89.19 |
99.18 |
|
10 |
Tetrahydrofuran |
87.99 |
98.78 |

a Reaction conditions: compound 2 (1 g), CAT05 (1%), HCOOH (1.1 equiv.), room temperature.
b It was detected by high-performance liquid chromatography employing an IC column.
Different hydrogen donors including ammonium formate (HCOONH4), sodium formate (HCOONa), and HCOOH were screened. As shown in [Table 4], when HCOONH4 and HCOONa were used as the hydrogen donors, no product was detected in this system, whereas, when HCOOH was used instead, compound 1 was obtained with 85.62% yield and 99.32% ee, thus, HCOOH was chosen as a hydrogen donor for the ATH reaction.
|
|
|||
|---|---|---|---|
|
Entry |
Hydrogen proton donor |
Yield (%)[d] |
ee (%)[e] |
|
1 |
HCOONH4 [a] |
ND |
/ |
|
2 |
HCOONa[b] |
ND |
/ |
|
3 |
HCOOH[c] |
85.62% |
99.32% |

Abbreviations: HCOONH4, ammonium formate; HCOONa, sodium formate; HCOOH, formic acid; ND, not detected.
a Compound 2 (0.503 g), CAT05 (0.5% equiv.), HCOONH4 (1.1 equiv.), room temperature, 23 hours.
b Compound 2 (0.501 g), CAT05 (1.0% equiv.), HCOONa (1.1 equiv.), room temperature, 17 hours.
c Compound 2 (2 g), CAT05 (0.5% equiv.), HCOOH (1.1 equiv.), 40°C, 15.5 hours.
d Detected by thin-layer chromatography.
e It was detected by high-performance liquid chromatography employing an IC column.
Conclusion
This work explored the synthesis route for compound 1 using pyridine-2,3-dicarboxylic acid (3) as a starting material. This robust process featured efficient, scalable, and stereoselective properties, meanwhile avoiding using dangerous reagents. The route afforded compound 1 in 92.1% yield with high purity (more than 99.8%) and 99.9% ee. The Ru-catalyzed ATH reaction was the key step of the route, and the optimal conditions for the reaction were: CAT05 (0.5%) as a catalyst, DIPEA as a base, dichloromethane as a solvent, and formic acid as a hydrogen donor. Moreover, it is successfully scaled up on the kilogram scale and this process is cost-saving which is suitable for further application.
Experimental Section
Reagents and Materials
Common commercially available materials were purchased from Xilong (Shantou, China), Aladdin (Shanghai, China), or Kelong (Chengdu, China). CAT01 was purchased from Sino Compound Catalysts Co., Ltd. (Suzhou, China). CAT02, CAT03, CAT04, and CAT05 were synthesized in-house. Unless otherwise stated, all the chemicals and reagents were commercially available and used without any depuration. Nuclear magnetic resonance (NMR) spectra were collected on a Bruker Avance 400 spectrometer in chloroform-d (CDCl3). Chemical shifts were reported in parts per million (ppm). The reference peak was defined as 7.26 ppm in the 1H spectrum and 77.0 ppm in 13C spectrum. Coupling constants are represented by Hz. High-resolution mass spectrometry was detected by a Fourier-transform ion cyclotron resonance mass spectrometer. Purity and ee values were detected by high-performance liquid chromatography using an Agilent Zorbax Eclipse XBridge C18 (250 mm × 4.6 mm; 5μm) and an IC column.
Dimethyl Pyridine-2,3-dicarboxylate (8)
To a 2 L round bottom flask was added compound 3 (50.5 g, 0.30 mol) and methanol (600 mL), followed by dropwise addition of concentrated sulfuric acid (45 mL) for 25 minutes under a nitrogen atmosphere. After stirring for an additional 10 minutes at room temperature, the mixture was heated to 75°C, stirred for 22.5 hours, and then cooled to room temperature. After the removal of methanol by distillation under vacuum at 41°C, the mixture was stirred at 0°C for 5 minutes followed by the addition of ice water and saturated NaHCO3 solution (650 mL) dropwise. Then, a large amount of white solid was precipitated. Ethyl acetate (300 mL) was added followed by the addition of saturated Na2CO3 solution (80 mL) to adjust pH = 8 to 9. The reaction mixture was poured into a separatory funnel and separated. The aqueous layer was extracted with EA (100 mL × 6). The organic layers were concentrated to afford an off-white solid (51.28 g), which was washed with n-hexane (150 mL) at room temperature for 2 hours and stirred at 0°C for 30 minutes, the solid was filtered and the filter cake was washed with 30 mL of n-hexane, dried in a vacuum at 38°C to give 9 (48.34 g, 81.97 yield%) as a white powder, which was used directly for the next reaction.
Dimethyl-5,9-dihydroxy-7H-cyclohepta[b]pyridine-6,8-dicarboxylate (9)
To a 1 L round bottom flask was added compound 9 (25.14 g, 128.8 mmol) and toluene (190 mL), followed by the addition of dimethyl glutarate (31.54 g, 196.9 mmol). A solution of potassium tert-butoxide (36.51 g, 325.37 mmol) in tetrahydrofuran (320 mL) was added to the reaction mixture dropwise at 0°C for 40 minutes. The mixture was stirred at room temperature for several minutes and allowed to stir for 2 hours at 58°C under a nitrogen atmosphere. After removal of the solvent by distillation under vacuum at 40°C, the mixture was added with ice water (400 mL) and ethyl acetate (250 mL), followed by the addition of glacial acetic acid dropwise to adjust pH = 7. The solution was poured into a separatory funnel and separated. The aqueous layer was extracted with ethyl acetate (200 mL, 100 mL × 8). The merged organic layers were washed with saturated NaCl solution (200 mL) and concentrated by distillation under vacuum at 40°C to afford a light-yellow solid, which was dispersed in n-hexane (100 mL), stirred at 0°C for 1 hour, and then filtered. The filter cake was washed with n-hexane (30 mL) and dried in a vacuum at 38°C to give 9 (25.98 g, 69.25 yield%) as a light-yellow powder.
7,8-Dihydro-5H-cyclohepta[b]pyridine-5,9(6H)-dione (2)
To a 1 L round bottom flask was added compound 9 (64.62 g, 0.22 mol), followed by the addition of 6 N HCl (312 mL, 1.87 mol). The reaction mixture was allowed to warm to 78°C and heat for 19.5 hours. After cooling to 0°C, saturated Na2CO3 solution (500 mL) was added carefully to adjust pH = 7 to precipitate solids. The mixture was poured into a separatory funnel, and extracted with dichloromethane (300 mL, 100 mL × 3). The dichloromethane layers were combined and then concentrated to dryness under reduced pressure to give the crude product. The crude product was added ethyl acetate (65 mL). The reaction liquid was warmed to 50°C and stirred till dissolved completely. Upon the addition of 150 mL of n-hexane, the reaction liquid was allowed to stir at 75°C for 0.5 hours. After standing layered, the reaction liquid was stirred at room temperature for 4 hours until a large amount of white solid precipitated. After stirring at 0°C for 1 hour, the slurry was filtered and the filter cake was washed with n-hexane (100 mL) and dried under reduced pressure at 38°C to afford 2 (34.81 g, 89.56 yield%) as an off-white powder.
(R)-9-Hydroxy-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-5-one (1)
To a 500 mL two-necked round bottom flask was added 2 (50.03 g, 285.6 mmol), CAT05 (783 mg, 1.1 mmol, 0.4% equiv.), and dichloromethane (400 mL), and the mixture was purged with N2 five times, then the mixture was stirred at room temperature. To the mixture was added DIPEA (15.2 g, 129.25 mmol), followed by portion-wise addition of formic acid (13.54 g, 293.98 mmol, 1.03) for 12 minutes. The reaction mixture was allowed to stir at room temperature (30°C) for 40 hours, concentrated to dryness under reduced pressure at 40°C. To the residue was added tert-butyl acetate (60 mL) and n-hexane (32 mL), and the mixture was stirred at 0°C. Upon solid precipitation, n-hexane (120 mL) was added, and the solution was stirred at 0°C for 30 minutes, and then the mixture was settled at room temperature. The slurry was filtrated under reduced pressure and the filter cake was washed with n-hexane (100 mL) to afford the crude product. The crude product was purified by silica gel chromatography to afford light-yellow liquid. The light-yellow liquid was recrystallized in n-hexane and ethyl acetate to give a white powder (41.76 g, 92.1% yield, 99.8% purity, and 99.9% ee). 1H-NMR (400 MHz, CDCl3) δ 8.67 (dd, J = 4.8, 1.4 Hz, 1H), 8.10 (dd, J = 7.7, 1.5 Hz, 1H), 7.38 (dd, J = 7.7, 4.9 Hz, 1H), 5.45 (s, 1H), 4.97 (dd, J = 9.7, 4.7 Hz, 1H), 3.04–2.67 (m, 2H), 2.56–2.39 (m, 1H), 2.12–1.96 (m, 1H), 1.89–1.70 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 202.9, 160.1, 150.3, 137.3, 132.1, 122.9, 70.7, 41.3, 33.3, 19.1.
Conflict of Interest
None declared.
-
References
- 1 Blair HA. Rimegepant: a review in the acute treatment and preventive treatment of migraine. CNS Drugs 2023; 37 (03) 255-265
- 2 Scott LJ. Rimegepant: first approval. Drugs 2020; 80 (07) 741-746
- 3 Wu YH, Wang J. Rimegepant (Nurtec ODT) [in Chinese]. Zhongguo Yaowu Huaxue Zazhi 2021; (02) 166
- 4 Holland PR, Goadsby PJ. Targeted CGRP small molecule antagonists for acute migraine therapy. Neurotherapeutics 2018; 15 (02) 304-312
- 5 Leahy DK, Fan Y, Desai LV. et al. Efficient and scalable enantioselective synthesis of a CGRP antagonist. Org Lett 2012; 14 (18) 4938-4941
- 6 Luo G, Chen L, Conway CM, Kostich W, Macor JE, Dubowchik GM. Asymmetric synthesis of heterocyclic analogues of a CGRP receptor antagonist for treating migraine. Org Lett 2015; 17 (24) 5982-5985
- 7 Li ZB, Huang H, Xia H, Qi XQ, Lin YF. Synthesis method for rimegepant by halogentaion reaction. CN Patent 115677694A. February, 2023
- 8 Ma YL, Jiao XC, Wang ZJ. Engineering a transaminase for the efficient synthesis of a key intermediate for rimegepant. Org Process Res Dev 2022; 26 (07) 1971-1977
- 9 Leahy DK, Yu F, Desai LV, Hanson RL, Rosner T, Luo G. CGRP Receptor Antagonists. U.S. Patent 20120010402A1. January, 2012
- 10 Luo G, Chen L, Conway CM. et al. Discovery of (5S,6S,9R)-5-amino-6-(2,3-difluorophenyl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-9-yl 4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxylate (BMS-927711): an oral calcitonin gene-related peptide (CGRP) antagonist in clinical trials for treating migraine. J Med Chem 2012; 55 (23) 10644-10651
- 11 Jones G, Jones RK. Annulation of pyridine as a route to quinolines, isoquinolines, and cyclo-heptapyridines. J Chem Soc, Perkin Trans 1973; I: 26-32
- 12 Luo GL. Piperidine derivatives as CGRP receptor antagonists. WO Patent 2009126530A2. October, 2019
- 13 Guo WC, Fang J, Wu HF, Wang GP. Industrial preparation for high optical purity remegipam intermediate. CN Patent 114805206A. July, 2022
- 14 Zhang J, Blazecka PG, Bruendl MM, Huang Y. Ru-TsDPEN with formic acid/Hunig's base for asymmetric transfer hydrogenation, a practical synthesis of optically enriched N-propyl pantolactam. J Org Chem 2009; 74 (03) 1411-1414
- 15 Zheng YP, Zhang XM, Zhang RT, Ma BD. Kilogram synthesis of (R)-(-)-denopamine by Ir/f-amphox catalyzed asymmetric hydrogenation. Green Synth Catal 2021; 2 (04) 393-396
- 16 Wang YZ, Hu L, Bai ST, Zhang X. Ru-catalyzed asymmetric reductive amination of aryl-trifluoromethyl ketones for synthesis of primary α-(trifluoromethyl)arylmethylamines. Org Lett 2023; 25 (27) 5033-5037
- 17 Yu JF, Huang FP, Fang W. et al. Discovery and development of ferrocene-based tetradentate ligands for Ir-catalysed asymmetric hydrogenation of ketone. Green Synth Catal 2022; 3 (02) 175-178
- 18 Hao W, Joe CL, Darù A. et al. Kinetic and thermodynamic considerations in the Rh-catalyzed enantioselective hydrogenation of 2-pyridyl-substituted alkenes. ACS Catal 2022; 12 (10) 5961-5969
- 19 Zhu LY, Cui YF, Chen X. et al. Synthesis of single stereoisomers of 2,2-disubstituted 3-hydroxycyclohexane-1-ones via enzymatic desymmetric reduction of the 1,3-cyclohexanediones. Green Synth Catal 2021; 2 (03) 320-323
- 20 Kempson J, Hou XP, Sun JH. et al. Synthesis optimization, scale-up, and catalyst screening efforts toward the MGAT2 clinical candidate, BMS-963272. Org Process Res Dev 2022; 26 (04) 1327-1335
- 21 Li BB, Zhang J, Chen FF, Chen Q, Xu JH, Zheng GW. Direct reductive amination of ketones with amines by reductive aminases. Green Synth Catal 2021; 2 (04) 345-349
Address for correspondence
Publication History
Received: 10 December 2023
Accepted: 30 January 2024
Article published online:
11 March 2024
© 2024. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Blair HA. Rimegepant: a review in the acute treatment and preventive treatment of migraine. CNS Drugs 2023; 37 (03) 255-265
- 2 Scott LJ. Rimegepant: first approval. Drugs 2020; 80 (07) 741-746
- 3 Wu YH, Wang J. Rimegepant (Nurtec ODT) [in Chinese]. Zhongguo Yaowu Huaxue Zazhi 2021; (02) 166
- 4 Holland PR, Goadsby PJ. Targeted CGRP small molecule antagonists for acute migraine therapy. Neurotherapeutics 2018; 15 (02) 304-312
- 5 Leahy DK, Fan Y, Desai LV. et al. Efficient and scalable enantioselective synthesis of a CGRP antagonist. Org Lett 2012; 14 (18) 4938-4941
- 6 Luo G, Chen L, Conway CM, Kostich W, Macor JE, Dubowchik GM. Asymmetric synthesis of heterocyclic analogues of a CGRP receptor antagonist for treating migraine. Org Lett 2015; 17 (24) 5982-5985
- 7 Li ZB, Huang H, Xia H, Qi XQ, Lin YF. Synthesis method for rimegepant by halogentaion reaction. CN Patent 115677694A. February, 2023
- 8 Ma YL, Jiao XC, Wang ZJ. Engineering a transaminase for the efficient synthesis of a key intermediate for rimegepant. Org Process Res Dev 2022; 26 (07) 1971-1977
- 9 Leahy DK, Yu F, Desai LV, Hanson RL, Rosner T, Luo G. CGRP Receptor Antagonists. U.S. Patent 20120010402A1. January, 2012
- 10 Luo G, Chen L, Conway CM. et al. Discovery of (5S,6S,9R)-5-amino-6-(2,3-difluorophenyl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-9-yl 4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxylate (BMS-927711): an oral calcitonin gene-related peptide (CGRP) antagonist in clinical trials for treating migraine. J Med Chem 2012; 55 (23) 10644-10651
- 11 Jones G, Jones RK. Annulation of pyridine as a route to quinolines, isoquinolines, and cyclo-heptapyridines. J Chem Soc, Perkin Trans 1973; I: 26-32
- 12 Luo GL. Piperidine derivatives as CGRP receptor antagonists. WO Patent 2009126530A2. October, 2019
- 13 Guo WC, Fang J, Wu HF, Wang GP. Industrial preparation for high optical purity remegipam intermediate. CN Patent 114805206A. July, 2022
- 14 Zhang J, Blazecka PG, Bruendl MM, Huang Y. Ru-TsDPEN with formic acid/Hunig's base for asymmetric transfer hydrogenation, a practical synthesis of optically enriched N-propyl pantolactam. J Org Chem 2009; 74 (03) 1411-1414
- 15 Zheng YP, Zhang XM, Zhang RT, Ma BD. Kilogram synthesis of (R)-(-)-denopamine by Ir/f-amphox catalyzed asymmetric hydrogenation. Green Synth Catal 2021; 2 (04) 393-396
- 16 Wang YZ, Hu L, Bai ST, Zhang X. Ru-catalyzed asymmetric reductive amination of aryl-trifluoromethyl ketones for synthesis of primary α-(trifluoromethyl)arylmethylamines. Org Lett 2023; 25 (27) 5033-5037
- 17 Yu JF, Huang FP, Fang W. et al. Discovery and development of ferrocene-based tetradentate ligands for Ir-catalysed asymmetric hydrogenation of ketone. Green Synth Catal 2022; 3 (02) 175-178
- 18 Hao W, Joe CL, Darù A. et al. Kinetic and thermodynamic considerations in the Rh-catalyzed enantioselective hydrogenation of 2-pyridyl-substituted alkenes. ACS Catal 2022; 12 (10) 5961-5969
- 19 Zhu LY, Cui YF, Chen X. et al. Synthesis of single stereoisomers of 2,2-disubstituted 3-hydroxycyclohexane-1-ones via enzymatic desymmetric reduction of the 1,3-cyclohexanediones. Green Synth Catal 2021; 2 (03) 320-323
- 20 Kempson J, Hou XP, Sun JH. et al. Synthesis optimization, scale-up, and catalyst screening efforts toward the MGAT2 clinical candidate, BMS-963272. Org Process Res Dev 2022; 26 (04) 1327-1335
- 21 Li BB, Zhang J, Chen FF, Chen Q, Xu JH, Zheng GW. Direct reductive amination of ketones with amines by reductive aminases. Green Synth Catal 2021; 2 (04) 345-349