

Subscribe to RSS
DOI: 10.1055/s-0044-1780496
Discovery of Topoisomerase I Inhibitor Nitidine Derivatives with IL-10 Enhancing Activity for the Treatment of Sepsis
Authors
Funding This work was supported by the Three-year Action Plan for Shanghai TCM Development and Inheritance Program (Grant No. ZY [2021-2023]-0401) and Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (Grant No. ZYYCXTD-D-202004).


Abstract
Nitidine chloride (NC) is a natural product that promotes the expression of interleukin-10 (IL-10) in macrophages by inhibiting topoisomerase I (TopoI) under stimulation by lipopolysaccharides (LPSs) and can be used in the treatment of sepsis. However, NC's poor water solubility limits its applications. This study aimed to design and synthesize a series of derivatives by simplifying the A- and E-rings in the structure of NC and introducing oxygen-containing groups, using NC as the lead compound. In this work, the ability of NC and its derivatives to induce IL-10 secretion and inhibit TopoI was evaluated. The water solubility of the compounds was determined in phosphate-buffered saline. An LPS-induced sepsis in mice was prepared to assess the activity of the compounds in vivo. Our data suggested that compound 6F showed better activity in inducing IL-10 secretion and inhibiting TopoI, and its water solubility was at least 500-fold higher than that of NC. When septic mice were given 6F (3 mg/kg), their survival rate was comparable to those treated with NC. Based on our findings, 6F may be a new drug candidate for the treatment of sepsis.
Introduction
Sepsis is a systemic inflammatory syndrome caused by the uncontrolled response of the organism to infection, which can lead to multiple organ dysfunction and death. It is currently one of the leading causes of death in patients in the surgical intensive care unit. It is also a common complication in patients suffering from severe burns that can lead to septic shock or death. Despite the recent advances in anti-infection treatment for severe sepsis or septic shock, wound management, as well as organ function support, the mortality rate due to sepsis is still high, ranging from 30 to 70%.[1] [2] [3] Excessive inflammatory response is a major cause of sepsis, and the release of pro-inflammatory factors ultimately causes circulatory dysfunction and the subsequent multi-organ failure, therefore, preventing excessive inflammatory response may be the focus of sepsis treatment.[4]
Interleukin-10 (IL-10) is reported to be an important anti-inflammatory factor that blocks the release of pro-inflammatory factors by activated monocytes.[5] [6] A single injection of IL-10 reduces the mortality of an animal model of sepsis, which was induced by lipopolysaccharide (LPS; also known as endotoxin) in mice.[7] [8] Intraperitoneal injection of IL-10 attenuates inflammatory response and inhibits the activation of the coagulation system and fibrinolysis in a human endotoxin model.[9] [10] Therefore, IL-10 is essential in the negative regulation of endotoxin-induced host responses.
Natural product nitidine chloride (NC; structure shown in [Fig. 1]), derived from Zanthoxylum nitidum (Roxb.) DC root, has anticancer, anti-inflammatory, antimalarial, and antibacterial properties.[11] [12] [13] [14] [15] [16] [17] In our previous study, we selected more than 500 monomeric compounds related to the development, differentiation, and functions of macrophages from the Chinese medicine compound library based on a screening platform.[11] We found that at a noncytotoxic dose (0–4 μmol/L), NC potentially prevented sepsis, and the underlying mechanisms may be the inhibition of TopoI, the induction of repairable DNA damage, and the subsequent activation of DNA damage response and the promotion of Akt activation. This synergistically enhances the activation of the PI3K–Akt pathway and facilitates the transcription and expression of IL-10 in response to LPS stimulation.[11]

However, the application of NC is limited due to its poor water solubility and low bioavailability.[12] [18] Water solubility is an essential physicochemical property of an organic small-molecule drug. It is also an important issue during the drug discovery. High water solubility often results in good drug potency and an ideal pharmacokinetic profile.[19] Theoretically, structural modification of a drug is an effective way to improve its water solubility, and this can be achieved by salt formation, introduction of polar groups, reduction of liposolubility, conformation optimization, as well as the incorporation of prodrugs. In this work, we attempted to simplify the structure of NC by removing the benzene ring to decrease its liposolubility and enhance its water solubility. We further assessed the activity of the NC derivatives obtained in increasing IL-10 and preventing lethality in an animal model of sepsis.
Results and Discussions
Synthesis
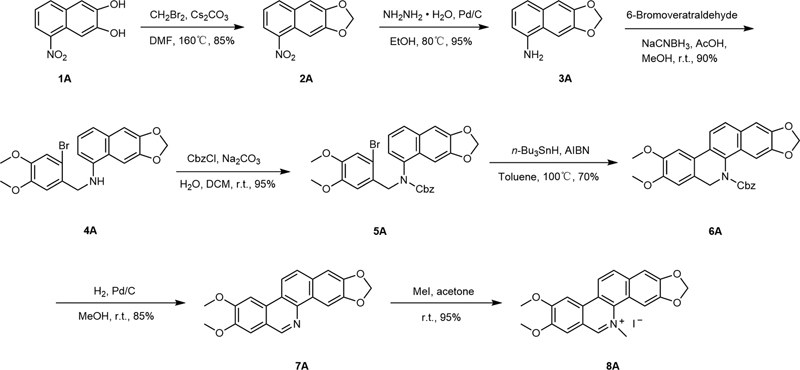
NC is abundant in plants; however, the total yield from natural sources is extremely low (0.003–0.07%) due to its limitation of poor solubility, making it expensive to obtain and unsuitable for extraction and production in large scale. In this work, a synthetic route for NC has been designed based on a retrosynthesis analysis and literature reviews ([Scheme 1]). The route started with 5-nitro-2,3-naphthalenediol (1A). The phenolic hydroxyl groups of 1A were protected with dibromomethane to give 2A, and the nitro group of which was reduced to an amino group using hydrazine hydrate and palladium carbon. Compound (3A) undergoes a reductive amination reaction with 6-bromoveratraldehyde, followed by the subsequent protection of the amino group using benzyloxycarbonyl (Cbz) to obtain the intermediate 5A, which gives a cyclization product 6A via a free-radical mechanism under the action of the free-radical initiators (n-Bu3SnH and azodiisobutyronitrile [AIBN]). After removing the Cbz-protecting group of compound 6A, the oxygen in the air during the posttreatment process oxidizes the carbon and nitrogen single bonds to a double bond to generate the intermediate 7A without any additional oxidation. In the presence of methyl iodide, a nitrogen methylation of 7A was achieved to give the target product (8A). The total yield of the route was approximately 40%.
NC belongs to the benzophenanthridine group of compounds, in which four aromatic rings, A, B, C, and D, are linked together to form a conjugated planar structure, which is highly hydrophobic. Reducing the number of aromatic rings in the NC structure may be an effective strategy to increase its water solubility. According to the existing structure–activity relationships, the two methoxyl groups at C-8 and C-9 should be left unchanged, the methylenedioxy (E-ring), piperonyl ring (A- and E-ring), and A-ring can be simplified to improve the druggability. Therefore, NC derivatives 6B, 6C, and 6D were designed and synthesized. Their synthetic pathway is similar to that of NC, and the starting materials include 4-amino-1,3-benzodioxol (1B), naphthylamine (1C), and aniline (1D), respectively ([Scheme 2]).

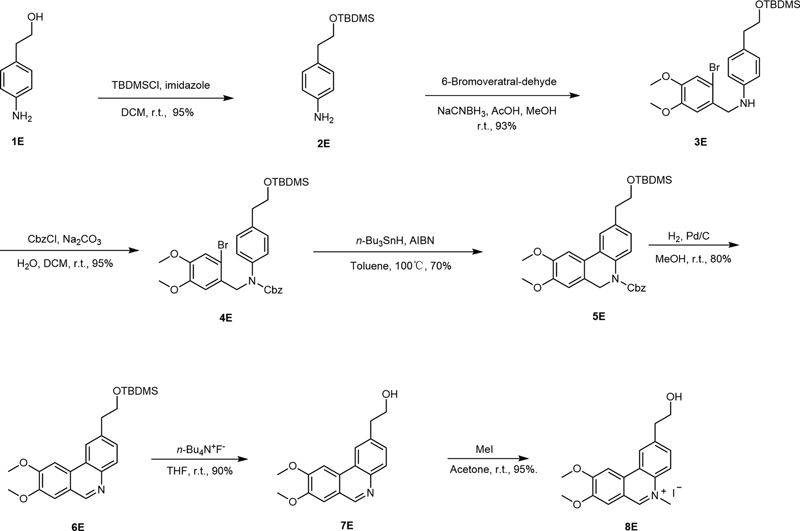
Oxygen-containing groups were introduced at the para-position of the N atom on 6D to improve the water solubility of the compound. The generated compounds included 6F (an ethyl carboxylate at the para-position of the N atom), 6H (an isopropoxy group at the para-position of the N atom), 7H (a phenolic hydroxyl at the para-position of the N atom), 8E (a hydroxyethyl group at the para-position of the N atom), and 9G (a hydroxymethyl group at the para-position of the N atom). The synthetic routes of 8E, 6F, and 6H are shown in [Schemes 3], [4], and [5], respectively, which are similar to those of NC, and the starting materials include p-aminophenethyl alcohol (1E), ethyl 4-aminobenzoate (1F), and 4-isopropoxy aniline (1H) with a total yield of 35 to 40%. [Scheme 4] shows the synthetic route of 9G. The ester group in compound 5F was reduced to hydroxyl by DIBAL-H, and then underwent Dess–Martin oxidation, N methylation, and sodium borohydride reduction to obtain the target product (9G). As shown in [Scheme 5], the isopropyl group in 6H was removed under the action of BCl3 to obtain 7H.


Evaluation of the IL-10 Secretion-Promoting Activity
The activity of the derivatives and some intermediates on macrophage RAW264.7 was assessed to screen compounds with better IL-10 secretion-promoting activity and to explore the structure–activity relationship of NC in promoting IL-10 secretion. Based on the formula (NC)% = (X – LPS)/(NC – LPS) ([Table 1]).[20] In the formula, X represents the average value of the three data sets obtained from the experimental group, where the LPS represents the average value of the control group in the three experiments, and NC represents the average value of the three experiments after treatment with LPS and NC. Unfortunately, the activity of IL-10 secretion of the selected derivatives and intermediates at a concentration of 2 μmol/L was weaker than that of NC. The activity of 8A on the secretion of IL-10 is approximately 65% of the NC, indicating that the anion may affect the secretion of IL-10. The activity of the derivatives and intermediates without A-ring (2B–6B), E-ring (2C–6C), and A- and E-rings (2D–6D) was also assessed with the maximum effect being seen in 6B (59.37%), 6C (57.08%), and 6D (46.92%), revealing that quaternary ammonium salt structures are important in promoting the secretion of IL-10. The A- and E-rings may be essential to maintain the IL-10 secretion-promoting activity and simplifying them will weaken their activities. In addition to simplifying the A- and E-rings, different oxygen-containing substituents at the para-position of N were designed. These compounds included ethyl carboxylate derivative (6E) and aldehyde derivative (8G), which showed better activity, as well as hydroxymethyl (9G) and isopropoxyl (6H), which were inactive, indicating that the oxygen-containing substituents affect the activity while the electron-withdrawing substituents might enhance the activity.
Abbreviation: LPS, lipopolysaccharide.
Note: Data were presented as average value ± standard deviation of three repeats.
The activity of a few derivatives with better activity was determined at 5 and 10 μmol/L. We found that when the concentration of NC and its derivatives were 5 and 10 μmol/L, their activities in promoting IL-10 secretion decreased compared with those at 2 μmol/L, suggesting better activities of the compound at lower concentrations. Meanwhile, fewer adherent and floating cells were found in the NC-treated group (5 μmol/L), which proved a stronger inhibitory effect of the compound on cell proliferation at the concentrations, subsequently leading to a decrease in the IL-10 secretion-promoting activity.
In vitro TopoI Inhibition Assay
NC promotes IL-10 secretion from macrophages by inhibiting TopoI activity, thus, inhibitory activity of NC and its derivatives for TopoI was preliminary screened at the concentration of 10 μmol/L. Given that, the derivatives containing quaternary ammonium salt structures facilitated the secretion of IL-10. These derivatives were further screened for TopoI inhibitory activity at a concentration of 5 μmol/L. TopoI inhibitors act by stabilizing a covalent TopoI–DNA complex called the cleavable complex. The ability of derivatives to stabilize this complex was evaluated by incubating TopoI and supercoiled DNA pBR322 in the presence of drugs. Cleavable complexes were revealed by the appearance of short DNA fragments when the samples were analyzed by gel electrophoresis under denaturing conditions, and the results were shown in [Fig. 2]. Our data showed that the positive control drugs camptothecin (CPT) and NC, as well as NC derivatives 8A, 6B, and 7H, completely inhibited TopoI-mediated DNA supercoil relaxation at a concentration of 5 μmol/L. Derivatives 6C and 6D had no TopoI inhibitory activity, indicating the important effect of the A- and E-ring structures in maintaining the activity. Derivatives 6F and 6H partially inhibited TopoI activity at a concentration of 5 μmol/L, while 8E and 9G had no activity. This indicated that the para-oxygen substituent of N affects the activity. Subsequently, the TopoI inhibitory activity of 8A, 6B, 6F, and 6H was re-screened at 2 μmol/L, of which only 6H showed no effect.

The mode of binding between the derivatives and TopoI was investigated. The derivatives were analyzed using docking analysis with the Schrodinger software (10.6) ([Fig. 3]). Arg364, Asp533, Asn722, and other critical amino acid residues were found at the active sites of TopoI.[21] [22] The 3D structure is shown in [Fig. 3A]. The docking result showed that the methoxy groups in the NC ([Fig. 3B]) and 6B ([Fig. 3C]) form hydrogen bonds with Asn722, and the methylenedioxy groups form hydrogen bonds with Lys425 and Arg364, respectively. The planar structure allows their insertion into the DNA cleavage site, forming base-stacking interactions with upstream and downstream base pairs. Therefore, their TopoI inhibitory activities are relatively strong, showing partial inhibitory activity at a concentration of 2 μmol/L. Although the hydroxymethyl group in 9G can form hydrogen bonds with Thr718 ([Fig. 3D]), it cannot form hydrogen bonds with other critical residues, such as Arg364 and Asn722, nor can it embed in the DNA cracks via a π–π interaction. Therefore, the TopoI inhibitory activity of 9G was weak, with no activity at a concentration of 5 μmol/L. This can partially explain the strength of the inhibitory activity of TopoI in the derivatives.

Derivatives with structures similar to NC display consistency in promoting IL-10 secretion and inhibiting TopoI activities. For example, 6B and 6F have strong activity on both IL-10 secretion and TopoI inhibition. This consistency was not observed in the derivatives with structures different from NC, as they might promote the secretion of IL-10 via other mechanisms. Based on these results, we summarized the relationship between derivative structure and their activity on IL-10 secretion and TopoI inhibition as follows:
-
The quaternary ammonium salt structure is essential for the pharmacological effects.
-
The A- and E-rings are important for maintaining activity.
-
The para-substituent of N influences the activity, for instance, phenolic hydroxyl groups and hydroxyethyl groups weaken the activity of IL-10 secretion and TopoI inhibition of the compound.
Water Solubility Determination
Water solubility of 6B and 6F was determined in phosphate-buffered saline (PBS). As presented in [Table 2], compared with NC, the solubility of 6B and 6F in PBS was improved to varying degrees, especially 6F (10,600 μg/mL) was more than 500-fold higher than NC (18.707 μg/mL). Although the water solubility of 6B is worse than 6F, it is also a factor of 7 higher than NC. This suggests that reducing the number of aromatic rings, removing the A-ring for example, and introducing hydrophilic groups in the structure of NC can improve the water solubility while maintaining the activity of the compound.
|
Compd. |
λmax (nm) |
Water solubility (μg/mL) |
|---|---|---|
|
NC |
272,295,330 |
18.707 |
|
6B |
298,352 |
139.168 |
|
6F |
275,350 |
>10,600 |
In vitro Activity
The in vivo activities of 6B and 6F in a mouse sepsis model were evaluated. As shown in [Fig. 4], NC (3 and 10 mg/kg) could significantly improve the survival rate in comparison to the model group, the survival rate of NC (10 mg/kg) reaching 80% within 84 hours. However, 6B (3 and 10 mg/kg) as well as 6F (10 mg/kg) has no significantly effect while 6F (3 mg/kg) significantly enhanced the survival rate of septic mice. Given above, 6F was preferred for the further research.

Conclusion
NC prevents sepsis by inhibiting TopoI activity and promoting IL-10 secretion by macrophages. However, its development has been limited due to its poor water solubility and low oral bioavailability. In this study, we simplified the core structure of the NC to obtain 6B, 6C, and 6D. Then, oxygen-containing substituents were introduced at the para-position of the N atom of compound 6D. The activity of the derivatives and intermediates in IL-10 secretion and TopoI inhibition were evaluated to elucidate the structure–activity relationship. Among them, derivatives 6B and 6F showed consistent in promoting IL-10 secretion and inhibiting TopoI activity. In addition, their water solubility was 7-fold and 500-fold higher than that of NC. Moreover, 6F (3 mg/kg) significantly improved the survival rate of septic mice, which was comparable to that of NC (3 mg/kg) within 84 hours. 6F can be used as a novel lead compound for further research. The study provides new strategies and drug candidates for discovery of TopoI inhibitors with better activity and druggability for the treatment of sepsis.
Experimental Section
Materials and Methods
Reagents (5-nitro-2,3-dihydroxynaphthalene, 6-bromoveratraldehyde, 4-amino-1,3-benzodioxol, naphthylamine, aniline, p-aminophenethyl alcohol, ethyl 4-aminobenzoate, and 4-isopropoxy aniline, etc.) were purchased from Alfa Aesar, Bidepharm, and Tokyo Chemical Industry and used directly without further processing, unless otherwise noted. Nuclear magnetic resonance (NMR) data were obtained using a Bruker DRX instrument in CDCl3 (chloroform-d) or DMSO-d 6 (methyl sulfoxide-d 6) at 500 MHz for 1H, and 125 Hz for 13C. The molecular weight and purity of the compounds were determined by Waters Corporation's separation module, LC/MS e2695. All reactions were monitored by thin-layer chromatography (TLC) using silica gel plates (silica gel 60 F254 0.25 mm).
Synthesis of 5-Nitronaphtho[2,3-d][1,3]dioxole (2A)
5-Nitro-2,3-dihydroxynaphthalene (1A) (2.05 g, 0.01 mol), dibromomethane (1.75 mL, 0.025 mol), and cesium carbonate (8.15 g, 0.025 mol) were suspended in N,N-dimethylformamide (50 mL). The reaction mixture was stirred at 160°C for 8 hours. After cooling to room temperature, the reaction was poured into a mixture of ethyl acetate (EA; 200 mL) and water (100 mL). The aqueous layer was removed. The organic layer was washed with water (100 mL) and brine (100 mL), dried over sodium sulfate, filtered, and concentrated to provide 2A (1.84 g, 85%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.10 (dd, J = 7.8, 1.2 Hz, 1H), 7.96 (s, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.36 (t, J = 7.9 Hz, 1H), 7.18 (s, 1H), 6.12 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 148.6, 146.2, 133.6, 132.4, 123.0, 122.9, 122.8, 104.5, 102.1, 100.3.
Synthesis of Naphtho[2,3-d][1,3]dioxol-5-amine (3A)
2A (2.17 g, 0.01 mol) was dissolved in ethanol (40 mL). The resulting solution was admixed with hydrazine hydrate (3.72 mL) and 10% palladium/carbon catalyst (383 mg) and heated under reflux for 2 hours. After cooling down to the room temperature, the catalyst was filtered out. The filtrate was concentrated to give 3A (1.77 g, 95%) as a white solid, which was used for the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 7.18 (s, 1H), 7.15 (d, J = 6.6 Hz, 1H), 7.13 (s, 1H), 7.09 (s, 1H), 6.69 (dd, J = 6.6, 1.8 Hz, 1H), 6.03 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 147.6, 147.3, 141.6, 131.6, 125.0, 120.2, 118.8, 109.6, 104.7, 101.1, 97.8; HR-ESI-MS (m/z): calcd. for C11H10NO2 + [M + H]+ 188.0706, found 188.0709.
Synthesis of N-(2-Bromo-4,5-dimethoxybenzyl)naphtho[2,3-d][1,3]dioxol-5-amine (4A)
A solution of 3A (2.45 g, 0.01 mol) and 6-bromoveratraldehyde (2.45 g, 0.01 mol) in methanol (20 mL) was added to acetic acid (1 mL). The resulting mixture was stirred for 1 hour. Then sodium cyanoborohydride (1.26 g, 0.02 mol) was added. After 8 hours, the solvent was removed under reduced pressure and extracted with dichloromethane (DCM; 20 mL × 3). The combined organic layers were washed with brine (10 mL), dried over sodium sulfate, filtered, and concentrated to obtain the white solid product 4A (3.74 g, 90%). 1H NMR (500 MHz, DMSO-d 6) δ 7.68 (s, 1H), 7.16 (d, J = 4.6 Hz, 2H), 7.06–7.01 (m, 2H), 6.97 (d, J = 8.0 Hz, 1H), 6.54 (s, 1H), 6.21 (d, J = 7.5 Hz, 1H), 6.10 (s, 2H), 4.37 (d, J = 5.7 Hz, 2H), 3.76 (s, 3H), 3.56 (s, 3H); 13C NMR (125 MHz, DMSO-d 6) δ 148.4, 148.3, 146.8, 146.4, 143.0, 131.0, 130.2, 124.9, 119.0, 115.8, 115.6, 112.4, 112.3, 104.1, 103.3, 100.9, 98.5, 55.9, 55.6, 46.8. HR-ESI-MS (m/z): calcd. for C20H19BrNO4 + [M + H]+ 416.0492, found 416.0499.
Synthesis of Benzyl(2-bromo-4,5-dimethoxybenzyl)(naphtho[2,3-d][1,3]dioxol-5-yl) carbamate (5A)
To a solution of 4A (700 mg, 1.68 mmol) and sodium carbonate (268 mg, 2.53 mmol) in DCM/H2O (12 mL/3 mL) was added benzyl chloroformate (0.48 mL, 3.37 mmol) dropwise. The reaction mixture was vigorously stirred at room temperature for 1 hour. After the completion of the reaction monitored by TLC, the mixture was extracted with DCM (20 mL × 3), washed with brine (10 mL), dried over sodium sulfate, filtered, and concentrated to give a crude, which was purified by silica gel column chromatography (petroleum ether [PE]:EA = 5–10%) to obtain 5A (876 mg, 95%) as a white solid. 1H NMR (500 MHz, DMSO-d6 ) δ 7.66 (d, J = 8.2 Hz, 1H), 7.47–6.92 (m, 10H), 6.83 (s, 1H), 6.12 (d, J = 5.0 Hz, 2H), 4.93 (dd, J = 169.1, 14.7 Hz, 4H), 3.69 (s, 3H), 3.51 (s, 3H); 13C NMR (125 MHz, DMSO-d6 ) δ 148.8, 148.1, 147.9, 147.4, 136.7, 131.2, 128.2, 128.2, 127.9, 127.6, 126.9, 124.0, 119.5, 115.2, 113.5, 104.0, 101.4, 98.8, 55.8, 55.3, 52.7. HR-ESI-MS (m/z): calcd. for C28H24BrNO6Na+ [M + Na]+ 572.0679, found 572.0678.
Synthesis of Benzyl-2,3-dimethoxy-[1,3]dioxolo[4',5′:4,5]benzo[1,2-c]phenanthridine-12(13H)-carboxylate (6A)
To a solution of 5A (624 mg, 1.00 mmol) and tri-n-butyltin hydride (538 μL, 2.00 mmol) in toluene was added AIBN (246 mg, 1.50 mmol in 5 mL toluene) dropwise under N2. The mixture was stirred at 100°C for 4 hours and cooled to room temperature. The toluene was removed under reduced pressure to give a crude, which was purified by silica gel column chromatography (PE:EA = 5–10%) to give 6A (328 mg, 70%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.68–7.60 (m, 2H), 7.34 (s, 1H), 7.30–6.95 (m, 7H), 6.86 (s, 1H), 6.05–6.00 (m, 2H), 5.30 (s, 2H), 5.20 (d, J = 12.4 Hz, 1H), 4.21 (d, J = 15.0 Hz, 1H), 3.99 (s, 3H), 3.94 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 149.0, 148.3, 147.7, 130.5, 128.4, 128.0, 126.1, 120.0, 116.2, 107.5, 104.1, 101.7, 101.3, 67.9, 56.4, 56.2; HR-ESI-MS (m/z): calcd. for C28H23NO6Na+ [M + Na]+ 492.1418, found 492.1414.
Synthesis of 2,3-Dimethoxy-[1,3]dioxolo[4',5′:4,5]benzo[1,2-c]phenanthridine (7A)
To a mixture of 6A (2.13 g, 4.54 mmol) in methanol (100 mL) in a Parr bottle was added 10% palladium/carbon catalyst (500 mg). Shake the slurry under 50 psi of H2 (g) pressure at room temperature overnight. The mixture was filtered through a pad of Celite and rinsed with DCM (100 mL × 2). The solution was concentrated to give 7A (1.28 g, 85%) as a yellow solid, which was used in the next step without further purification. 1H NMR (500 MHz, DMSO-d 6) δ 9.30 (s, 1H), 8.62 (d, J = 9.1 Hz, 1H), 8.54 (s, 1H), 8.15 (s, 1H), 7.94 (d, J = 8.9 Hz, 1H), 7.69 (s, 1H), 7.50 (s, 1H), 6.19 (s, 2H), 4.07 (s, 3H), 3.97 (s, 3H); 13C NMR (125 MHz, DMSO-d 6) δ 153.5, 149.3, 148.4, 146.3, 140.7, 140.4, 129.7, 128.7, 126.7, 122.3, 119.9, 108.1, 104.9, 103.4, 101.5,101.3, 56.5, 56.3. HR-ESI-MS (m/z): calcd. for C20H16NO4 + [M + H]+ 334.1074, found 334.1086.
Synthesis of Nitidine (8A)
7A (1.0 g, 3.00 mmol) was dissolved in methyl iodide (5 mL) and heated in a sealed tube for 6 hours. The precipitate was collected, washed with ether (20 mL), and concentrated to provide 8A (1.35 g, 95%) as a yellow solid. 1H NMR (500 MHz, methanol-d4 ) δ 9.63 (s, 1H), 8.75 (d, J = 8.9 Hz, 1H), 8.29 (s, 1H), 8.22 (d, J = 11.1 Hz, 2H), 7.81 (s, 1H), 7.59 (s, 1H), 6.27 (s, 2H), 4.94 (s, 3H), 4.26 (s, 3H), 4.11 (s, 3H); 13C NMR (125 MHz, methanol-d4 ) δ 170.7, 152.3, 150.1, 149.0, 133.1, 130.2, 120.0, 118.4, 108.0, 105.6, 103.4, 102.9, 102.6, 56.3, 55.6, 50.6. HR-ESI-MS (m/z): calcd. for C21H18NO4 + [M]+ 348.1230, found 348.1243.
Synthesis of N-(2-Bromo-4,5-dimethoxybenzyl)benzo[d][1,3]dioxol-4-amine (2B)
The synthetic process is similar to compound 4A. Yield: 90%. 1H NMR (500 MHz, CDCl3) δ 7.02 (s, 1H), 6.94 (s, 1H), 6.72–6.66 (m, 1H), 6.33 (dd, J = 7.8, 1.1 Hz, 1H), 6.28–6.22 (m, 1H), 5.90 (t, J = 0.9 Hz, 2H), 4.36 (s, 2H), 4.05 (s, 1H), 3.85 (s, 3H), 3.78 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 148.7, 148.5, 147.3, 133.9, 132.5, 130.2, 122.4, 115.6, 113.1, 112.2, 107.1, 100.6, 99.6, 56.2, 56.0, 48.3; HR-ESI-MS (m/z): calcd. for C16H17BrNO4 + [M + H]+ 366.0335, found 366.0335.
Synthesis of Benzylbenzo[d][1,3]dioxol-4-yl(2-bromo-4,5-dimethoxybenzyl) carbamate (3B)
3B was synthesized similar to that of 5A. Yield: 95%. 1H NMR (500 MHz, CDCl3) δ 7.29 (s, 5H), 6.93 (s, 1H), 6.90 (s, 1H), 6.71 (dt, J = 15.6, 7.8 Hz, 2H), 6.61 (s, 1H), 5.80 (s, 2H), 5.20 (s, 2H), 4.93 (s, 2H), 3.80 (s, 3H), 3.70 (s, 3H); 13C NMR (125MHz, CDCl3) δ 148.5, 148.3, 142.7, 128.4, 128.2, 127.8, 121.5, 120.7, 115.1, 107.3, 101.0, 67.5, 56.0, 55.7; HR-ESI-MS (m/z): calcd. for C24H22BrNO6Na+ [M + Na]+ 522.0523, found 522.0547.
Synthesis of Benzyl-7,8-dimethoxy-[1,3]dioxolo[4,5-c]phenanthridine-4(5H) carboxylate (4B)
4B was synthesized similar to that of 6A. Yield: 69%. HR-ESI-MS (m/z): calcd. for C24H21NO6Na+ [M + Na]+ 442.1261, found 442.1263.
Synthesis of 7,8-Dimethoxy-[1,3]dioxolo[4, 5-c]phenanthridine (5B)
5B was synthesized similar to that of 7A. Yield: 86%. 1H NMR (500 MHz, CDCl3) δ 9.06 (s, 1H), 7.95 (d, J = 8.7 Hz, 1H), 7.75 (s, 1H), 7.31 (s, 1H), 7.26–7.24 (d, 1H), 6.26 (s, 2H), 4.12 (s, 3H), 4.05 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 153.3, 152.5, 149.6, 146.4, 142.7, 130.7, 128.6, 121.0, 120.1, 115.0, 109.4, 108.5, 102.6, 102.0, 56.3, 56.3; HR-ESI-MS (m/z): calcd. for C16H14NO4 + [M + H]+ 284.0917, found 284.0919.
Synthesis of 7,8-Dimethoxy-4-methyl-[1,3]dioxolo[4,5-c]phenanthridin-4-ium iodide (6B)
6B was synthesized similar to that of 8A. Yield: 93%. 1H NMR (500 MHz, DMSO-d 6) δ 9.73 (s, 1H), 8.74 (d, J = 9.0 Hz, 1H), 8.26 (s, 1H), 7.82–7.76 (m, 2H), 6.41 (s, 2H), 4.61 (s, 3H), 4.19 (s, 3H), 4.00 (s, 3H); 13C NMR (125 MHz, DMSO-d 6) δ 158.5, 152.7, 150.6, 149.2, 137.7, 132.1, 121.0, 120.4, 119.4, 117.8, 112.2, 110.2, 103.6, 103.2, 57.3, 56.2, 48.1; HR-ESI-MS (m/z): calcd. for C17H16NO4 + [M]+ 298.1074, found 298.1061.
Synthesis of N-(2-Bromo-4,5-dimethoxybenzyl)naphthalen-1-amine (2C)
2C was synthesized similar to that of 4A. Yield: 89%. 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 8.4 Hz, 1H), 7.83–7.80 (m, 1H), 7.49–7.44 (m, 2H), 7.35–7.27 (m, 2H), 7.26 (s, 1H), 7.08 (s, 1H), 7.01 (s, 1H), 6.61 (d, J = 7.2 Hz, 1H), 4.51 (s, 2H), 3.89 (s, 3H), 3.75 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 149.0, 148.8, 134.4, 128.9, 126.7, 125.9, 125.0, 123.6, 120.0, 115.8, 113.5, 112.6, 56.4, 56.2, 48.9; HR-ESI-MS (m/z): calcd. for C19H19BrNO2 + [M + H]+ 372.0594, found 372.0599.
Synthesis of Benzyl(2-bromo-4,5-dimethoxybenzyl)(naphthalen-1-yl) (3C)
3C was synthesized similar to that of 5A. Yield: 96%. 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 9.0 Hz, 1H), 7.79 (d, J = 8.3 Hz, 1H), 7.74 (d, J = 6.8 Hz, 1H), 7.52–7.32 (m, 4H), 7.21–6.83 (m, 7H), 5.34 (d, J = 14.6 Hz, 1H), 5.09 (d, J = 25.4 Hz, 2H), 4.74 (d, J = 14.8 Hz, 1H), 3.80 (s, 3H), 3.65 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 156.5, 149.0, 148.5, 137.2, 136.8, 134.6, 130.8, 129.0, 128.5, 128.3, 127.8, 127.5, 126.8, 126.2, 126.1, 125.5, 122.7, 115.2, 113.4, 67.4, 56.2, 56.0; HR-ESI-MS (m/z): calcd. for C27H24BrNO4Na+ [M + Na]+ 528.0781, found 528.0785.
Synthesis of Benzyl-8,9-dimethoxybenzo[c]phenanthridine-5(6H)-carboxylate (4C)
4C was synthesized similar to that of 6A. Yield: 69%. 1H NMR (500 MHz, pyridine-d 5) δ 7.66 (s, 1H), 7.42 (d, J = 86.0 Hz, 6H), 7.20–6.91 (m, 5H), 6.53 (s, 1H), 5.49 (s, 2H), 4.93 (s, 2H), 3.70 (s, 3H), 3.62 (s, 3H).
Synthesis of 8,9-Dimethoxybenzo[c]phenanthridine (5C)
5C was synthesized similar to that of 7A. Yield: 85%. 1H NMR (500 MHz, CDCl3) δ 9.37 (d, J = 8.3 Hz, 1H), 9.30 (s, 1H), 8.39 (d, J = 9.1 Hz, 1H), 7.96 (dd, J = 8.4, 3.1 Hz, 2H), 7.89 (s, 1H), 7.76 (ddd, J = 8.3, 6.9, 1.3 Hz, 1H), 7.67 (ddd, J = 8.1, 6.9, 1.3 Hz, 1H), 7.40 (s, 1H), 4.16 (s, 3H), 4.09 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 153.2, 150.1, 150.0, 141.0, 133.0, 132.4, 128.9, 127.8, 127.5, 127.2, 127.1, 124.6, 122.8, 120.8, 119.9, 107.4, 101.8, 56.3, 56.3; HR-ESI-MS (m/z): calcd. for C19H16NO2 + [M + H]+ 290.1176, found 290.1179.
Synthesis of 8,9-Dimethoxy-5-methylbenzo[c]phenanthridin-5-ium iodide (6C)
6C was synthesized similar to that of 8A. Yield: 93%. 1H NMR (500 MHz, DMSO-d 6) δ 9.58 (s, 1H), 9.07 (d, J = 8.2 Hz, 1H), 8.88 (d, J = 9.1 Hz, 1H), 8.30–8.19 (m, 3H), 7.98 (s, 1H), 7.86 (dt, J = 26.5, 7.3 Hz, 2H), 4.18 (s, 3H), 4.02 (s, 3H), 3.38 (s, 3H); HR-ESI-MS (m/z): calcd. for C20H18NO2 + [M]+ 304.1332, found 304.1339.
Synthesis of N-(2-Bromo-4,5-dimethoxybenzyl)aniline (2D)
2D was synthesized similar to that of 4A. Yield: 89%. 1H NMR (500 MHz, CDCl3) δ 7.18 (dd, J = 8.5, 7.4 Hz, 2H), 7.04 (s, 1H), 6.95 (s, 1H), 6.74 (td, J = 7.4, 1.0 Hz, 1H), 6.64 (dd, J = 8.6, 1.0 Hz, 2H), 4.33 (s, 2H), 3.87 (s, 3H), 3.78 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 148.8, 148.7, 148.0, 130.4, 129.4, 115.8, 113.3, 113.2, 112.4, 56.4, 56.2, 48.5; HR-ESI-MS (m/z): calcd. for C15H17BrNO2 + [M + H]+ 322.0437, found 322.0432.
Synthesis of Benzyl(2-bromo-4,5-dimethoxybenzyl)(phenyl)carbamate (3D)
3D was synthesized similar to that of 5A. Yield: 96%. 1H NMR (500 MHz, DMSO-d6 ) δ 7.34–7.18 (m, 10H), 7.05 (s, 1H), 6.80 (s, 1H), 5.15 (s, 2H), 4.89 (s, 2H), 3.72 (s, 3H), 3.56 (s, 3H); 13C NMR (125 MHz, DMSO-d6 ) δ 154.7, 148.6, 148.1, 136.7, 128.7, 128.3, 127.9, 127.8, 127.4, 127.0, 126.5, 115.5, 112.5, 66.7, 55.8, 55.4, 52.7; HR-ESI-MS (m/z): calcd. for C23H22BrNO4Na+ [M + Na]+ 478.0624, found 478.0626.
Synthesis of Benzyl 8,9-dimethoxyphenanthridine-5(6H)-carboxylate (4D)
4D was synthesized similar to that of 6A. Yield: 70%. 1H MR (500 MHz, acetone-d 6) δ 7.82 (dd, J = 7.5, 1.6 Hz, 1H), 7.63 (d, J = 7.1 Hz, 1H), 7.43–7.28 (m, 6H), 7.23 (dtd, J = 16.4, 7.3, 1.5 Hz, 2H), 6.99 (s, 1H), 5.21 (s, 2H), 4.78 (s, 2H), 3.90 (s, 3H), 3.86 (s, 3H); 13C NMR (125 MHz, acetone-d6 ) δ 154.2, 150.6, 150.4, 137.6, 129.8, 129.3, 128.8, 128.8, 128.0, 127.5, 125.9, 125.7, 125.1, 124.3, 110.4, 108.3, 68.1, 56.4, 56.2, 47.3; HR-ESI-MS (m/z): calcd. for C23H21NO4Na+ [M + Na]+ 398.1363, found 398.1368.
Synthesis of 8,9-Dimethoxyphenanthridine (5D)
5D was synthesized similar to that of 7A. Yield: 86%. 1H NMR (500 MHz, CDCl3) δ 9.14 (s, 1H), 8.43 (d, J = 8.1 Hz, 1H), 8.16 (d, J = 8.2 Hz, 1H), 7.87 (s, 1H), 7.71–7.61 (m, 2H), 7.34 (s, 1H), 4.13 (s, 3H), 4.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 153.1, 151.8, 150.1, 144.0, 130.2, 128.3, 127.9, 126.7, 123.9, 121.8, 107.9, 101.9, 56.3, 56.2; HR-ESI-MS (m/z): calcd. for C15H14NO2 + [M + H]+ 240.1019, found 240.1027.
Synthesis of 8,9-Dimethoxy-5-methylphenanthridin-5-ium iodide (6D)
6D was synthesized similar to that of 8A. Yield: 94%. 1H NMR (500 MHz, DMSO-d6 ) δ 9.90 (s, 1H), 9.19–9.15 (m, 1H), 8.46–8.39 (m, 2H), 8.07 (dt, J = 24.5, 7.5 Hz, 2H), 7.90 (s, 1H), 4.58 (s, 3H), 4.22 (s, 3H), 4.03 (s, 3H); 13C NMR (125 MHz, DMSO-d6 ) δ 158.2, 151.7, 151.2, 133.5, 131.8, 131.2, 129.3, 124.8, 124.5, 119.6, 119.0, 110.2, 103.6, 57.3, 56.3, 45.3; HR-ESI-MS (m/z): calcd. for C16H16NO2 + [M]+ 254.1176, found 254.1171.
Synthesis of 4-(2-((tert-Butyldimethylsilyl)oxy)ethyl)aniline (2E)
To a solution of 2-(4-aminophenyl)ethan-1-ol (0.68 g, 5.00 mmol) in DCM (6 mL) was added imidazole (0.48 g, 6.00 mmol) and tert-butyldimethylsilyl chloride (0.90 g, 6.00 mmol). The reaction mixture was stirred at room temperature for 4 hours. After the completion of the reaction monitored by TLC, the reaction was quenched with water (5 mL) at 0°C and extracted with DCM (20 mL × 2). The organic phases were combined and dried over sodium sulfate, concentrated under reduced pressure to obtain 2E (1.17 g, 94%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.01 (d, J = 8.1 Hz, 2H), 6.63 (d, J = 8.4 Hz, 2H), 3.76 (t, J = 7.3 Hz, 2H), 2.73 (t, J = 7.3 Hz, 2H), 0.91 (s, 9H), 0.02 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 144.6, 130.0, 129.2, 115.3, 65.1, 38.9, 26.1, 18.5, -5.2; HR-ESI-MS (m/z): calcd. for C14H26NOSi+ [M + H]+ 252.1778, found 252.1772.
Synthesis of N-(2-Bromo-4,5-dimethoxybenzyl)-4-(2-((tert-butyldimethylsilyl)oxy)ethyl)ani-line (3E)
3E was synthesized similar to that of 4A. Yield: 89%. 1H NMR (500 MHz, CDCl3) δ 7.03 (s, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.95 (s, 1H), 6.57 (d, J = 8.5 Hz, 2H), 4.30 (s, 2H), 3.86 (s, 3H), 3.78 (s, 3H), 3.74 (t, J = 7.3 Hz, 2H), 2.71 (t, J = 7.3 Hz, 2H), 0.88 (s, 9H), 0.00 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 148.8, 148.7, 146.3, 130.5, 130.0, 128.6, 115.7, 113.3, 113.2, 112.4, 65.1, 56.3, 56.2, 48.8, 38.9, 26.1, 18.5, -5.2; HR-ESI-MS (m/z): calcd. for C23H35BrNO3Si+ [M + H]+ 480.1564, found 480.1569.
Synthesis of Benzyl(2-bromo-4,5-dimethoxybenzyl)(4-(2-((tert-butyldimethylsilyl)oxy)ethyl) phenyl) carbamate (4E)
4E was synthesized similar to that of 5A. Yield: 96%. 1H NMR (500 MHz, acetone-d 6) δ 7.33 (d, J = 4.6 Hz, 5H), 7.18 (s, 4H), 7.01 (s, 1H), 6.91 (s, 1H), 5.18 (s, 2H), 4.95 (s, 2H), 3.81–3.78 (m, 5H), 3.62 (s, 3H), 2.76 (d, J = 6.6 Hz, 2H), 0.83 (s, 9H), -0.07 (s, 6H); 13C NMR (125 MHz, acetone-d 6) δ 156.1, 150.2, 149.9, 138.7, 138.1, 130.4, 129.6, 129.2, 128.7, 128.6, 127.5, 116.5, 113.4, 67.7, 64.8, 56.4, 56.1, 54.0, 39.6, 26.3, 18.8, -5.3; HR-ESI-MS (m/z): calcd. for C31H40BrNO5SiNa+ [M + Na]+ 636.1751, found 636.1757.
Synthesis of Benzyl 2-(2-((tert-butyldimethylsilyl)oxy)ethyl)-8,9-dimethoxyphenanthridine-5 (6H)-carboxylate (5E)
5E was synthesized similar to that of 6A. Yield: 70%. 1H NMR (500 MHz, CDCl3) δ 7.51 (s, 1H), 7.35 (d, J = 4.2 Hz, 5H), 7.26 (s, 1H), 7.23 (s, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.78 (s, 1H), 5.21 (s, 2H), 4.76 (s, 2H), 3.97 (s, 3H), 3.92 (s, 3H), 3.86 (t, J = 6.8 Hz, 2H), 2.86 (d, J = 6.8 Hz, 2H), 0.87 (s, 9H), −0.01 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 149.1, 136.4, 128.6, 128.2, 128.1, 127.9, 124.8, 124.1, 109.1, 106.8, 67.8, 64.6, 56.4, 56.2, 47.0, 39.5, 26.1, -5.2; HR-ESI-MS (m/z): calcd. for C31H39NO5SiNa+ [M + Na]+ 556.2490, found 556.2488.
Synthesis of 2-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-8,9-dimethoxyphenanthridine (6E)
6E was synthesized similar to that of 7A. Yield: 86%. 1H NMR (500 MHz, CDCl3) δ 9.10 (s, 1H), 8.25 (s, 1H), 8.07 (d, J = 8.3 Hz, 1H), 7.86 (s, 1H), 7.54 (dd, J = 8.4, 1.7 Hz, 1H), 7.34 (s, 1H), 4.14 (s, 3H), 4.06 (s, 3H), 3.94 (t, J = 6.6 Hz, 2H), 3.08 (t, J = 6.6 Hz, 2H), 0.86 (s, 9H), -0.05 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 153.0, 151.0, 150.0, 142.6, 138.2, 129.8, 129.6, 128.2, 123.7, 122.1, 110.1, 107.9, 101.9, 64.5, 56.3, 56.2, 40.0, 26.1, 18.4, -5.3; HR-ESI-MS (m/z): calcd. for C23H32NO3Si+ [M + H]+ 398.2146, found 398.2140.
Synthesis of 2-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-8,9-dimethoxy-5-methylphenanthridin-5-ium (7E)
To a solution of 6E (397 mg, 1.00 mmol) in tetrahydrofuran (5 mL) was added tetrabutylammonium fluoride (313 mg, 1.20 mmol). The mixture was stirred at room temperature for 0.5 hours and quenched by the addition of ammonium chloride solution. Removed tetrahydrofuran under a reduced pressure. Extracted the resulting mixture with EA (10 mL ×2). The organic phases were combined, dried over sodium sulfate, concentrated to provide a crude solid, which was purified by silica gel column chromatography (PE:EA = 40–50%) to give 7E (266 mg, 94%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.95 (s, 1H), 8.19 (s, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.75 (s, 1H), 7.52 (dd, J = 8.4, 1.7 Hz, 1H), 7.21 (s, 1H), 4.12 (s, 3H), 4.05 (d, J = 7.3 Hz, 5H), 3.13 (t, J = 6.3 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 152.9, 151.2, 150.0, 142.6, 137.5, 130.3, 129.1, 127.9, 123.8, 122.0, 107.8, 101.8, 63.7, 56.4, 56.2, 39.8; HR-ESI-MS (m/z): calcd. for C17H18NO3 + [M + H]+ 284.1281, found 284.1287.
Synthesis of 2-(2-Hydroxyethyl)-8,9-dimethoxy-5-methylphenanthridin-5-ium iodide (8E)
8E was synthesized similar to that of 8A. Yield: 90%. 1H NMR (500 MHz, DMSO-d 6) δ 9.78 (s, 1H), 8.94 (s, 1H), 8.37–8.31 (m, 2H), 7.96 (d, J = 8.7 Hz, 1H), 7.85 (s, 1H), 4.54 (s, 3H), 4.21 (s, 3H), 4.02 (s, 3H), 3.82 (t, J = 6.6 Hz, 2H), 3.11 (t, J = 6.5 Hz, 2H), 2.12 (s, 1H); 13C NMR (125 MHz, DMSO-d 6) δ 158.0, 151.1, 150.8, 141.9, 132.5, 132.1, 131.5, 124.5, 124.2, 119.2, 118.9, 110.1, 103.4, 61.8, 57.3, 56.2, 52.8, 45.2; HR-ESI-MS (m/z): calcd. for C18H20NO3 + [M]+ 298.1438, found 298.1427.
Synthesis of Ethyl 4-((2-bromo-4,5-dimethoxybenzyl)amino)benzoate (2F)
2F was synthesized similar to that of 4A. Yield: 86%. 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 8.8 Hz, 2H), 7.04 (s, 1H), 6.87 (s, 1H), 6.59 (d, J = 8.8 Hz, 2H), 4.37 (s, 2H), 4.31 (q, J = 7.1 Hz, 2H), 3.86 (s, 3H), 3.77 (s, 3H), 1.35 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.9, 151.5, 149.1, 148.8, 131.6, 129.3, 119.6, 115.9, 113.3, 112.2, 112.0, 60.4, 56.4, 56.2, 47.9, 14.6; HR-ESI-MS (m/z): calcd. for C18H21BrNO4 + [M + H]+ 394.0648, found 394.0648.
Synthesis of Ethyl 4-(((benzyloxy)carbonyl)(2-bromo-4,5-dimethoxybenzyl)amino)benzoate (3F)
3F was synthesized similar to that of 5A. Yield: 95%. 1H NMR (500 MHz, acetone-d 6) δ 7.95 (d, J = 8.8 Hz, 2H), 7.44 (d, J = 8.7 Hz, 2H), 7.33 (d, J = 4.3 Hz, 5H), 7.03 (s, 1H), 6.90 (s, 1H), 5.22 (s, 2H), 5.04 (s, 2H), 4.31 (q, J = 7.1 Hz, 2H), 3.78 (s, 3H), 3.61 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, acetone-d 6) δ 166.1, 155.6, 150.3, 149.9, 137.6, 130.6, 129.2, 129.1, 128.9, 128.8, 128.7, 127.5, 127.3, 127.2, 116.6, 113.4, 113.4, 68.2, 61.5, 56.4, 56.2, 53.6, 14.6; HR-ESI-MS (m/z): calcd. for C26H26BrNO6Na+ [M + Na]+ 550.0836, found 550.0820.
Synthesis of 5-Benzyl 2-ethyl 8,9-dimethoxyphenanthridine-2,5(6H)-dicarboxylate (4F)
4F was synthesized similar to that of 6A. Yield: 67%. 1H NMR (500 MHz, acetone-d 6) δ 8.38 (d, J = 1.9 Hz, 1H), 7.87 (dd, J = 8.5, 1.9 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.46 (s, 1H), 7.44–7.31 (m, 5H), 7.01 (s, 1H), 5.25 (s, 2H), 4.83 (s, 2H), 4.37 (q, J = 7.1 Hz, 2H), 3.93 (s, 3H), 3.87 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, acetone-d 6) δ 166.4, 154.0, 151.1, 150.6, 137.3, 129.6, 129.3, 129.0, 128.9, 128.3, 127.8, 127.8, 125.4, 125.3, 124.2, 110.4, 108.3, 68.5, 61.5, 56.5, 56.3, 47.2, 14.6; HR-ESI-MS (m/z): calcd. for C26H25NO6Na+ [M + Na]+ 470.1574, found 470.1563.
Synthesis of Ethyl 8,9-dimethoxyphenanthridine-2-carboxylate (5F)
5F was synthesized similar to that of 7A. Yield: 89%. 1H NMR (500 MHz, CDCl3) δ 9.15 (d, J = 22.2 Hz, 2H), 8.26 (d, J = 6.8 Hz, 1H), 8.15 (d, J = 8.6 Hz, 1H), 7.91 (s, 1H), 7.34 (s, 1H), 4.49 (q, J = 7.1 Hz, 2H), 4.16 (s, 3H), 4.06 (s, 3H), 1.47 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.8, 153.8, 153.6, 150.5, 146.3, 130.2, 128.7, 128.3, 127.7, 124.7, 123.5, 122.1, 108.0, 102.1, 61.5, 56.6, 56.3, 14.6; HR-ESI-MS (m/z): calcd. for C18H18NO4 + [M + H]+ 312.1230, found 312.1238.
Synthesis of 2-(Ethoxycarbonyl)-8,9-dimethoxy-5-methylphenanthridin-5-ium iodide (6F)
6F was synthesized similar to that of 8A. Yield: 90%. 1H NMR (500 MHz, DMSO-d 6) δ 9.99 (s, 1H), 9.55 (s, 1H), 8.56 (d, J = 9.0 Hz, 1H), 8.48 (d, J = 7.0 Hz, 2H), 7.94 (s, 1H), 4.60 (s, 3H), 4.50 (q, J = 7.1 Hz, 2H), 4.28 (s, 3H), 4.04 (s, 3H), 1.44 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, DMSO-d 6) δ 164.8, 158.6, 153.4, 151.5, 135.8, 131.9, 130.3, 130.2, 126.1, 124.4, 120.5, 119.3, 110.5, 104.1, 61.8, 57.5, 56.4, 45.6, 14.2; HR-ESI-MS (m/z): calcd. for C19H20NO4 + [M]+ 326.1387, found 326.1401.
Synthesis of (8,9-Dimethoxyphenanthridin-2-yl)methanol (6G)
To a solution of 5F (311 mg, 1.00 mmol) in anhydrous DCM (10 mL) was added 1.0 mol/L diisobutylaluminum hydride (1.10 mL, solution in hexanes) dropwise at 0°C. The reaction mixture was warmed to room temperature and allowed to stir for 2 hours. The reaction was quenched by the addition of ammonium chloride solution, filtered through a pad of Celite, rinsed with DCM (10 mL ×2), washed with brine, dried over sodium sulfate, and concentrated to provide a crude, which was purified by silica gel column chromatography (PE:EA = 30–40%) to give 6G (185 mg, 69%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 9.08 (s, 1H), 8.40 (s, 1H), 8.11 (d, J = 8.4 Hz, 1H), 7.84 (s, 1H), 7.64 (dd, J = 8.4, 1.7 Hz, 1H), 7.31 (s, 1H), 4.97 (s, 2H), 4.13 (s, 3H), 4.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 153.1, 151.8, 150.2, 143.6, 139.4, 130.4, 128.3, 126.9, 123.9, 122.0, 119.8, 107.9, 102.0, 65.5, 56.4, 56.3; HR-ESI-MS (m/z): calcd. for C16H16NO3 + [M + H]+ 270.1125, found 270.1135.
Synthesis of 8,9-Dimethoxyphenanthridine-2-carbaldehyde (7G)
To a solution of 6G (269 mg, 1.00 mmol) in anhydrous DCM (5 mL) was added Dess–Martin periodinane (508 mg, 1.20 mmol) at 0°C. The resulting reaction mixture was stirred at room temperature for 2 hours and monitored by TLC. Upon completion, the reaction was neutralized with sodium sulfite (aq) and sodium bicarbonate (aq) at 0°C and extracted with DCM (10 mL × 2). The organic phases were combined, dried over sodium sulfate, and concentrated to give 7G (227 mg, 85%) as a yellow solid, which was directly used without further purification. HR-ESI-MS (m/z): calcd. for C16H14NO3 + [M + H]+ 268.0968, found 268.0977.
Synthesis of 2-Formyl-8,9-dimethoxy-5-methylphenanthridin-5-ium iodide (8G)
8G was synthesized similar to that of 8A. Yield: 80%. 1H NMR (500 MHz, pyridine-d 5) δ 10.42 (s, 1H), 10.29 (s, 1H), 9.80–9.78 (m, 1H), 8.64 (d, J = 8.9 Hz, 1H), 8.60 (dd, J = 8.9, 1.4 Hz, 1H), 8.56 (s, 1H), 8.04 (s, 1H), 4.90 (s, 3H), 4.16 (s, 3H), 4.05 (s, 3H); HR-ESI-MS (m/z): calcd. for C17H16NO3 + [M]+ 282.1125, found 282.1140.
Synthesis of 2-(Hydroxymethyl)-8,9-dimethoxy-5-methylphenanthridin-5-ium (9G)
To a solution of 8G (408 mg, 1.00 mmol) in tetrahydrofuran (10 mL) was added sodium borohydride (45.4 mg, 1.20 mmol) 0°C. Then the reaction mixture was warmed to room temperature and allowed to stir for 1 hour. The reaction was quenched by ammonium chloride solution, extracted with EA (10 mL × 2), washed with brine (10 mL), dried over sodium sulfate, filtered, and concentrated to give 9G (368 mg, 90%) as a yellow solid. 1H NMR (500 MHz, pyridine-d 5) δ 8.16 (s, 1H), 7.59 (s, 1H), 7.53 (d, J = 5.7 Hz, 2H), 6.85 (d, J = 8.2 Hz, 1H), 6.80 (s, 1H), 5.03 (s, 3H), 4.13 (s, 2H), 3.78 (d, J = 10.5 Hz, 6H); HR-ESI-MS (m/z): calcd. for C17H18NO3 + [M]+ 284.1281, found 284.1297.
Synthesis of N-(2-Bromo-4,5-dimethoxybenzyl)-4-isopropoxyaniline (2H)
2H was synthesized similar to that of 4A. Yield: 91%. 1H NMR (500 MHz, CDCl3) δ 7.03 (s, 1H), 6.95 (s, 1H), 6.77 (d, J = 8.9 Hz, 2H), 6.58 (d, J = 8.9 Hz, 2H), 4.36 (dq, J = 12.1, 6.1 Hz, 1H), 4.27 (s, 2H), 3.86 (s, 3H), 3.78 (s, 3H), 1.28 (d, J = 6.1 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 150.6, 148.7, 148.6, 142.3, 130.6, 118.0, 115.7, 114.6, 113.2, 112.4, 71.2, 56.3, 56.1, 49.4, 22.3; HR-ESI-MS (m/z): calcd. for C18H23BrNO3 + [M + H]+ 380.0856, found 380.0881.
Synthesis of Benzyl(2-bromo-4,5-dimethoxybenzyl)(4-isopropoxyphenyl)carbamate (3H)
3H was synthesized similar to that of 5A. Yield: 97%. 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 11.1 Hz, 5H), 6.92 (s, 3H), 6.78 (d, J = 8.8 Hz, 3H), 5.18 (s, 2H), 4.89 (s, 2H), 4.49 (hept, J = 6.0 Hz, 1H), 3.83 (s, 3H), 3.55 (d, J = 85.0 Hz, 3H), 1.31 (d, J = 6.1 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 156.5, 156.0, 148.8, 148.6, 138.5, 136.9, 129.0, 128.5, 128.0, 128.0, 116.1, 115.4, 70.2, 67.4, 56.2, 56.0, 53.9, 22.1; HR-ESI-MS (m/z): calcd. for C26H28BrNO5Na+ [M + Na]+ 536.1043, found 536.1040.
Synthesis of Benzyl 2-isopropoxy-8,9-dimethoxyphenanthridine-5(6H)-carboxylate (4H)
4H was synthesized similar to that of 6A. Yield: 69%. 1H NMR (500 MHz, CDCl3) δ 7.35 (s, 6H), 7.25–7.16 (m, 2H), 6.85–6.74 (m, 2H), 5.20 (s, 2H), 4.76 (s, 2H), 4.60 (hept, J = 6.0 Hz, 1H), 3.95 (s, 3H), 3.91 (s, 3H), 1.38 (d, J = 6.1 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 155.4, 149.2, 149.0, 136.5, 130.2, 128.6, 128.2, 128.0, 124.6, 113.9, 113.4, 111.3, 109.1, 107.0, 70.5, 67.8, 56.4, 56.2, 47.1, 22.3; HR-ESI-MS (m/z): calcd. for C26H27NO5Na+ [M + Na]+ 456.1781, found 456.1780.
Synthesis of 2-Isopropoxy-8,9-dimethoxyphenanthridine (5H)
5H was synthesized similar to that of 7A. Yield: 87%. 1H NMR (500 MHz, CDCl3) δ 8.98 (s, 1H), 8.06 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 2.6 Hz, 1H), 7.73 (s, 1H), 7.34–7.28 (m, 2H), 4.78 (p, J = 6.1 Hz, 1H), 4.10 (s, 3H), 4.04 (s, 3H), 1.43 (d, J = 6.1 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 156.5, 152.7, 150.1, 149.5, 139.2, 131.5, 127.6, 125.1, 122.0, 118.2, 107.8, 106.4, 102.0, 70.6, 56.3, 56.2, 22.3; HR-ESI-MS (m/z): calcd. for C18H20NO3 + [M + H]+ 298.1438, found 298.1438.
Synthesis of 2-Isopropoxy-8,9-dimethoxy-5-methylphenanthridin-5-ium iodide (6H)
6H was synthesized similar to that of 8A. Yield: 90%. 1H NMR (500 MHz, DMSO-d 6) δ 9.72 (s, 1H), 8.44 (d, J = 2.3 Hz, 1H), 8.39–8.29 (m, 2H), 7.86 (s, 1H), 7.73 (dd, J = 9.5, 2.6 Hz, 1H), 5.16 (hept, J = 6.0 Hz, 1H), 4.55 (s, 3H), 4.22 (s, 3H), 4.02 (s, 3H), 1.40 (d, J = 6.0 Hz, 6H); 13C NMR (125 MHz, DMSO-d 6) δ 157.9, 157.6, 151.2, 149.1, 130.9, 128.2, 126.6, 121.5, 120.9, 119.0, 110.1, 108.1, 103.9, 70.4, 57.4, 56.2, 21.7; HR-ESI-MS (m/z): calcd. for C19H22NO3 + [M]+ 312.1594, found 312.1606.
Synthesis of 2-Hydroxy-8,9-dimethoxy-5-methylphenanthridin-5-ium (7H)
To a solution of 6H (130 mg, 0.417 mmol) in anhydrous DCM (10 mL) was added boron trichloride (198 μL, 1.25 mmol) dropwise at –30 °C. The reaction mixture was warmed to room temperature and allowed to stir for 1 hour. The reaction mixture was quenched by the addition of ammonium chloride solution, extracted with DCM (10 mL × 2), washed with brine (10 mL), dried over sodium sulfate, filtered, and concentrated to give 7H (100 mg, 85%) as a yellow solid. 1H NMR (500 MHz, methanol-d 4) δ 8.78 (s, 1H), 7.78 (d, J = 9.4 Hz, 1H), 7.59 (s, 1H), 7.41 (s, 1H), 7.29 (d, J = 2.5 Hz, 1H), 7.20 (dd, J = 9.4 Hz, 2.5, 1H), 4.30 (s, 3H), 4.10 (s, 3H), 4.02 (s, 3H). 13C NMR (125 MHz, methanol-d4 ) δ 171.1, 158.3, 152.5, 143.9, 131.5, 129.1, 128.1, 125.6, 120.5, 120.4, 109.9, 108.7, 103.3, 57.2, 56.7, 45.5; HR-ESI-MS (m/z): calcd. for C16H16NO3 + [M]+ 270.1125, found 270.1132.
IL-10 Secretion Activity Assay
Cell Culture
A mouse macrophage cell line RAW264.7 was purchased from the Cell Bank of the Shanghai Institute of Biochemistry & Cell Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences (Shanghai, China, http://www.cellbank.org.cn). The cells were cultured in DMEM medium supplemented with 10% (v/v) fetal bovine saline in a humidified incubator (Thermo Scientific) in a 5% CO2 atmosphere at 37°C.
Enzyme-Linked Immunosorbent Assay
RAW264.7 cell suspension (2.5 × 105 cells/mL, 250 μL) was added into each well of a 48-well plate and incubated at 37°C with 5% CO2 for 24 hours. The test samples or an equivalent volume of DMSO (blank group) was added to the cells and incubated for another 24 hours followed by the addition of LPS (1 μg/mL) for 12 hours. The supernatant in each group was centrifuged at 3,000 rpm at 4°C for 5 minutes for the enzyme-linked immunosorbent assay (ELISA) according to the kit instructions (eBioscience, United States). In addition to the groups of the test compound and DMSO control, group C (without LPS stimulation) was set up for each experiment. There are three replicates in each group.
TopoI Inhibitory Activity Assay
TopoI inhibitory activity assay was conducted according to a reported study.[11] The compound was dissolved in DMSO with a final concentration of 10, 5, and 2 μmol/L, respectively. The compound at a specific concentration was mixed with 10 × DNA TopoI buffer (2 μL), 0.1% bovine serum albumin (2 μL), TopoI (0.5 U), pBR322 plasmid DNA (0.25 μg), and distilled water (varied as needed to bring the final volume to 20 μL) to achieve a final volume of 20 μL. The reactions were carried out for 15 minutes at 37°C and stopped by the addition of 2 μL of loading buffer × 10. The samples were electrophoresed on a 0.8% agarose gel in TAE (Tris-acetate-EDTA) running buffer at 120 V for 40 minutes and then stained with 0.5 μg/mL of ethidium bromide for 10 minutes. DNA bands were visualized using a UV transilluminator (Syngene G:BOX F3, England).
Computational Protocols for Protein Preparation
Receptor Preparation
The receptor structures were prepared using Schrödinger 2018 (Schrödinger, United States) by importing Topo I-DNA and norindenoisoquinoline crystal structures (PDB:1TL8) and analyzing the protein structure using the Protein Preparation Wizard, removing water molecules and redundant structures, as well as hydrogenation, side chain hydroxylation, side chain repair, and main chain end processing. Using the Receptor Grid Generation module, the active cavity was defined by the ligand norindenoisoquinoline in the crystal complex, and the docking radius was set to 10 Å.
Ligand Preparation
For docking analysis, NC and its derivatives (6B and 9G) were imported into Schrodinger in an SDF format. These small molecules were conformationally optimized using Ligand Prepare and Minimize modules.
Molecular Docking
The receptor active cavity, as defined earlier, and the derivatives were docked using Glid-Ligand docking, with XP (extra precision) selected for docking precision and flexible mode for ligand.
Water Solubility Determination
The excess compounds were dissolved in 200 μL of PBS, respectively, sonicated, and allowed to stand at room temperature for 24 hours, then filtered through a microporous filter membrane (0.45 μm). Next, each of the drug solution (100 μL) was diluted to 1.5 mL with PBS, and 10 μL of which was injected into the column for high-performance liquid chromatography analysis, and the peak area was recorded. An Agilent 1100 HPLC system (Agilent Technologies, MA, United States), equipped with a quaternary pump, a vacuum degasser, an autosampler, and a column heater-cooler, was used. Separation was performed by an Agilent Zorbax extend-C18 column (250 mm × 4.6 mm i.d., 5um, Agilent, United States). The column temperature was maintained at 25°C. Solvent A (water with 0.2% (v/v) acetic acid) and solvent B (acetonitrile) were used for gradient elution with the program as follows: 5 – 95% B (0 – 10 minutes); 95–95% B (10 – 15 minutes). The flow rate was set at 0.8 mL/min. The solubility data of each compound in PBS were calculated by an external standard method.
Animals
Male BALB/c mice (6- to 8-week-old) were obtained from Shanghai Laboratory Animal Company (Shanghai, China) and housed in the Experimental Animal House at the Second Military Medical University (Shanghai, China) in environmentally controlled conditions (22°C, a 12-hour light/dark cycle with the light cycle from 6:00 to 18:00 and the dark cycle from 18:00 to 6:00) with ad libitum access to standard laboratory chow. The study protocol was approved by the local institutional review board at the affiliated institutions. The animal experiments were conducted according to the established institutional guidelines for animal care and use at the Second Military Medical University.
Treatment
NC and its derivatives 6B and 6F were weighed (10 and 3 mg, respectively) and dissolved in 200 μL DMSO. Then, 5 mL of normal saline was added, and the mixture was sonicated for 20 minutes to dissolve the compounds completely. The mixture was diluted to 10 mL with normal saline to obtain sample solutions at 10 and 3 mg/kg doses.
Male BALB/c mice were randomly divided into eight groups, including a control, a model, and treatment groups. Equal volumes of normal saline and NC, 6B, and 6F (3 and 10 mg/kg) were injected intraperitoneally into the mouse for 6 hours before modeling. An experimental mouse model of sepsis was established by intraperitoneally injecting LPS (15 mg/kg). Then, the mice in each group were returned to the cages with free access to food and water. The number of dead mice was recorded every 12 hours, and the dead mice were removed. The survival rate was observed within 84 hours.
Statistical Analysis
The software of Graphpad Prism 8 was used to draw graphs and analyze data, and a log-rank test was used to analyze the differences in Kaplan–Meier survival curves. The results are expressed as mean ± standard deviation (mean ± SD); p ≤ 0.05 was considered statistically significant.
Conflict of Interest
None declared.
Supporting Information
Spectral data (1H NMR and 13C NMR) for compounds 2A–6A, 2B, 3B, 5B, 6B, 2C, 3C, 5C, 6C, 2D–6D, 2E–8E, 2F–6F, 6G, 8G, 9G, and 2H–7H are included in this Supporting Information ([Figs. S1–S39]) (available in the online version).
# These authors contributed equally to this work.
-
References
- 1 Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol 2004; 4 (02) 133-142
- 2 Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29 (07) 1303-1310
- 3 Coopersmith CM, Wunsch H, Fink MP. et al. A comparison of critical care research funding and the financial burden of critical illness in the United States. Crit Care Med 2012; 40 (04) 1072-1079
- 4 Zhao HQ, Li WM, Lu ZQ, Sheng ZY, Yao YM. The growing spectrum of anti-inflammatory interleukins and their potential roles in the development of sepsis. J Interferon Cytokine Res 2015; 35 (04) 242-251
- 5 de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med 1991; 174 (05) 1209-1220
- 6 Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol 2016; 197 (05) 1539-1546
- 7 Gérard C, Bruyns C, Marchant A. et al. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J Exp Med 1993; 177 (02) 547-550
- 8 Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med 1993; 177 (04) 1205-1208
- 9 Pajkrt D, Camoglio L, Tiel-van Buul MC. et al. Attenuation of proinflammatory response by recombinant human IL-10 in human endotoxemia: effect of timing of recombinant human IL-10 administration. J Immunol 1997; 158 (08) 3971-3977
- 10 Pajkrt D, van der Poll T, Levi M. et al. Interleukin-10 inhibits activation of coagulation and fibrinolysis during human endotoxemia. Blood 1997; 89 (08) 2701-2705
- 11 Yang N, Yue R, Ma J. et al. Nitidine chloride exerts anti-inflammatory action by targeting Topoisomerase I and enhancing IL-10 production. Pharmacol Res 2019; 148: 104368
- 12 Lu Q, Luo S, Shi Z, Yu M, Guo W, Li C. Nitidine chloride, a benzophenanthridine alkaloid from Zanthoxylum nitidum (Roxb.) DC., exerts multiple beneficial properties, especially in tumors and inflammation-related diseases. Front Pharmacol 2022; 13: 1046402
- 13 Cui Y, Wu L, Cao R. et al. Antitumor functions and mechanisms of nitidine chloride in human cancers. J Cancer 2020; 11 (05) 1250-1256
- 14 Wu YL, Liu X, Liu KL, Cui XL, Wang CF. Effects of nitidine chloride on ulcerative colitis in mice and its mechanism [in Chinese]. Chung Kuo Ying Yung Sheng Li Hsueh Tsa Chih 2019; 35 (06) 525-529
- 15 Bouquet J, Rivaud M, Chevalley S, Deharo E, Jullian V, Valentin A. Biological activities of nitidine, a potential anti-malarial lead compound. Malar J 2012; 11: 67
- 16 Shen C, Kuang Y, Xu S. et al. Nitidine chloride inhibits fibroblast like synoviocytes-mediated rheumatoid synovial inflammation and joint destruction by targeting KCNH1. Int Immunopharmacol 2021; 101 (Pt A): 108273
- 17 Liu Q, Wang T, Zhou L. et al. Nitidine chloride prevents OVX-induced bone loss via suppressing NFATc1-mediated osteoclast differentiation. Sci Rep 2016; 6: 36662
- 18 Chen YW, Liu HG, Yang L. et al. Study on equilibrium solubility and oil-water distribution coefficient of nitidine chloride. Lishizhen Med Mat Med Res 2010; 21 (05) 1121-1122
- 19 Li Z, Wang J, Zhou Y, Liu H. Lead compound optimization strategy (3)–Structure modification strategies for improving water solubility [in Chinese]. Yao Xue Xue Bao 2014; 49 (09) 1238-1247
- 20 Sundberg TB, Choi HG, Song JH. et al. Small-molecule screening identifies inhibition of salt-inducible kinases as a therapeutic strategy to enhance immunoregulatory functions of dendritic cells. Proc Natl Acad Sci U S A 2014; 111 (34) 12468-12473
- 21 Staker BL, Feese MD, Cushman M. et al. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem 2005; 48 (07) 2336-2345
- 22 Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin Jr AB, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A 2002; 99 (24) 15387-15392
Address for correspondence
Publication History
Received: 13 October 2023
Accepted: 29 January 2024
Article published online:
07 March 2024
© 2024. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol 2004; 4 (02) 133-142
- 2 Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29 (07) 1303-1310
- 3 Coopersmith CM, Wunsch H, Fink MP. et al. A comparison of critical care research funding and the financial burden of critical illness in the United States. Crit Care Med 2012; 40 (04) 1072-1079
- 4 Zhao HQ, Li WM, Lu ZQ, Sheng ZY, Yao YM. The growing spectrum of anti-inflammatory interleukins and their potential roles in the development of sepsis. J Interferon Cytokine Res 2015; 35 (04) 242-251
- 5 de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med 1991; 174 (05) 1209-1220
- 6 Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol 2016; 197 (05) 1539-1546
- 7 Gérard C, Bruyns C, Marchant A. et al. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J Exp Med 1993; 177 (02) 547-550
- 8 Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med 1993; 177 (04) 1205-1208
- 9 Pajkrt D, Camoglio L, Tiel-van Buul MC. et al. Attenuation of proinflammatory response by recombinant human IL-10 in human endotoxemia: effect of timing of recombinant human IL-10 administration. J Immunol 1997; 158 (08) 3971-3977
- 10 Pajkrt D, van der Poll T, Levi M. et al. Interleukin-10 inhibits activation of coagulation and fibrinolysis during human endotoxemia. Blood 1997; 89 (08) 2701-2705
- 11 Yang N, Yue R, Ma J. et al. Nitidine chloride exerts anti-inflammatory action by targeting Topoisomerase I and enhancing IL-10 production. Pharmacol Res 2019; 148: 104368
- 12 Lu Q, Luo S, Shi Z, Yu M, Guo W, Li C. Nitidine chloride, a benzophenanthridine alkaloid from Zanthoxylum nitidum (Roxb.) DC., exerts multiple beneficial properties, especially in tumors and inflammation-related diseases. Front Pharmacol 2022; 13: 1046402
- 13 Cui Y, Wu L, Cao R. et al. Antitumor functions and mechanisms of nitidine chloride in human cancers. J Cancer 2020; 11 (05) 1250-1256
- 14 Wu YL, Liu X, Liu KL, Cui XL, Wang CF. Effects of nitidine chloride on ulcerative colitis in mice and its mechanism [in Chinese]. Chung Kuo Ying Yung Sheng Li Hsueh Tsa Chih 2019; 35 (06) 525-529
- 15 Bouquet J, Rivaud M, Chevalley S, Deharo E, Jullian V, Valentin A. Biological activities of nitidine, a potential anti-malarial lead compound. Malar J 2012; 11: 67
- 16 Shen C, Kuang Y, Xu S. et al. Nitidine chloride inhibits fibroblast like synoviocytes-mediated rheumatoid synovial inflammation and joint destruction by targeting KCNH1. Int Immunopharmacol 2021; 101 (Pt A): 108273
- 17 Liu Q, Wang T, Zhou L. et al. Nitidine chloride prevents OVX-induced bone loss via suppressing NFATc1-mediated osteoclast differentiation. Sci Rep 2016; 6: 36662
- 18 Chen YW, Liu HG, Yang L. et al. Study on equilibrium solubility and oil-water distribution coefficient of nitidine chloride. Lishizhen Med Mat Med Res 2010; 21 (05) 1121-1122
- 19 Li Z, Wang J, Zhou Y, Liu H. Lead compound optimization strategy (3)–Structure modification strategies for improving water solubility [in Chinese]. Yao Xue Xue Bao 2014; 49 (09) 1238-1247
- 20 Sundberg TB, Choi HG, Song JH. et al. Small-molecule screening identifies inhibition of salt-inducible kinases as a therapeutic strategy to enhance immunoregulatory functions of dendritic cells. Proc Natl Acad Sci U S A 2014; 111 (34) 12468-12473
- 21 Staker BL, Feese MD, Cushman M. et al. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem 2005; 48 (07) 2336-2345
- 22 Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin Jr AB, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A 2002; 99 (24) 15387-15392