Keywords
α2-antiplasmin - plasmin inhibitor - diabetes
Introduction
Despite advances in therapies, vascular occlusive disease remains a major cause of both mortality and morbidity worldwide.[1] The risk of vascular thrombosis increases in conditions characterized by insulin resistance and deranged glucose metabolism.[2] Following external vessel injury, the cellular and acellular components of coagulation are activated to form a thrombus and limit blood loss, a normal physiological response. However, this becomes a pathological process when it occurs following internal vessel injury, such as rupture of an atheromatous plaque, leading to vessel occlusion and consequently resulting in organ damage.[3] Following vessel injury, platelets attach to the site of vascular lesion, become activated, and aggregate to help in thrombus formation while also contributing to the activation of coagulation factors. Tissue factor released following vessel injury starts a cascade of reactions that culminate in the formation of thrombin that converts soluble fibrinogen into the insoluble fibrin network, which forms the skeleton of the thrombus. When the fibrin network forms, the fibrinolytic system is activated to limit thrombus extension, a process controlled by a fine balance between fibrinolytic and antifibrinolytic proteins, highlighting the intricacy of the system.[4]
While several studies have shown the association between fibrin network characteristics and vascular disease,[5]
[6]
[7] it was not until relatively recently that a direct interaction between fibrin clot lysis and clinical outcome(s) was shown.[8]
[9] Therefore, targeting fibrin clot lysis is a legitimate strategy to reduce thrombosis risk, particularly in hypofibrinolytic states such as diabetes. Plasmin inhibitor (PI) is a key antifibrinolytic protein that is cross-linked into fibrin networks by activated factor (F) XIII. Therefore, reduction in PI concentrations in blood clots represents a credible approach to improve fibrinolysis, consequently resulting in reduced thrombosis risk.
In this review, we discuss the role of PI as an antifibrinolytic protein, focusing on mechanistic pathways, role of this protein in hypofibrinolytic states, and possible approaches to modulate protein function, to reduce thrombosis risk. Our literature search strategy is detailed in [Supplementary Material S1] (available in the online version). All titles and abstracts were screened, and only relevant articles written in English with full text provided were then selected.
Plasmin Inhibitor a Key Antifibrinolytic Protein
Plasmin Inhibitor a Key Antifibrinolytic Protein
Plasmin inhibitor is termed PI (or serpin F2) and was first discovered in 1976 independently by three groups and was referred to as antiplasmin, α2-PI, or primary PI.[10]
[11]
[12] For the purpose of this review, we will adhere to the term “PI” throughout. PI is a direct inhibitor of plasmin by forming plasmin–antiplasmin (PAP) complexes, unlike other antifibrinolytic proteins that exert an indirect effect.[4]
Biochemical Structure and Synthesis
PI is a 464-amino-acid-long glycoprotein with a molecular weight of approximately 70 kDa and contains atypical N- and C-terminal sequences flanking the serpin domain ([Fig. 1]).[13]
[14] The concentration of PI in the blood is roughly 1 µM (70 µg mL−1),[11]
[15] and it is produced mainly in the liver and also by the kidney and brain.[11]
[15]
[16]
[17] PI is the primary inhibitor of plasmin but also has the ability to inhibit neutrophil elastase, trypsin, and activated protein C.[18]
[19]
[20]
[21]
[22] PI has a half-life of approximately 60 hours, but plasmin–PI complexes are cleared with a half-life of 12 hours.[18]
 Fig. 1 Schematic representation of diversity forms of PI complexed with plasmin. PI includes a native form, which is 464 residues in length with N-terminal methionine (Met-PB-PI) and a truncated form, following digestion of the peptide bond between P12 and N13, leading to 12-residue shorter form (Asn-PB-PI). The N-terminal Q14-residue of PI is cross-linked to K303 in the Aα-chains of fibrinogen, and this process is performed by antiplasmin-cleaving enzyme (ACE) and catalyzed by activated coagulation factor XIII in the presence of Ca2+ ions. The C-terminal portion of antiplasmin contains a 51-residue extension with a secondary binding site that recognizes the lysine-binding sites of plasminogen and plasmin. PI, plasmin inhibitor.
Fig. 1 Schematic representation of diversity forms of PI complexed with plasmin. PI includes a native form, which is 464 residues in length with N-terminal methionine (Met-PB-PI) and a truncated form, following digestion of the peptide bond between P12 and N13, leading to 12-residue shorter form (Asn-PB-PI). The N-terminal Q14-residue of PI is cross-linked to K303 in the Aα-chains of fibrinogen, and this process is performed by antiplasmin-cleaving enzyme (ACE) and catalyzed by activated coagulation factor XIII in the presence of Ca2+ ions. The C-terminal portion of antiplasmin contains a 51-residue extension with a secondary binding site that recognizes the lysine-binding sites of plasminogen and plasmin. PI, plasmin inhibitor.
Different Plasmin Inhibitor Variants and Physiological Roles
PI undergoes multiple posttranslational modifications (PTMs), such as a sulphation of Y457[23]
[24] and multiple N-linked glycosylations at residues N99, N268, N282, and N289.[23]
[24] PI also undergoes proteolytic processing at both N- and C-termini, explaining the diversity of circulating PI isoforms or variants ([Fig. 2]).[24]
 Fig. 2 Diversity of circulating PI terminus and their physiological effects. PI is expressed and released by the liver and the kidney as a single-chain plasmin-binding protein with a methionine (Met) residue at the N-terminus (Met-PB-PI). Posttranslational modifications of N-terminal (blue box) and C-terminal (yellow box) result in several circulating PI forms. About 30% of PI circulates in plasma in the native. The remaining 70% of circulating PI is N-terminally cleaved between the proline (P) residue and the asparagine (N) residue and results in the truncated form Asn-PI, which is more effective at cross-linking. The C-terminal is found in plasma in two forms of which 70% is the full-length active form (Met-PB-PI) and 30% inactive (Met-NPB-PI). Only the active form can bind to plasmin(ogen) via a two-step process which can either be reversible or irreversible. PI, plasmin inhibitor.
Fig. 2 Diversity of circulating PI terminus and their physiological effects. PI is expressed and released by the liver and the kidney as a single-chain plasmin-binding protein with a methionine (Met) residue at the N-terminus (Met-PB-PI). Posttranslational modifications of N-terminal (blue box) and C-terminal (yellow box) result in several circulating PI forms. About 30% of PI circulates in plasma in the native. The remaining 70% of circulating PI is N-terminally cleaved between the proline (P) residue and the asparagine (N) residue and results in the truncated form Asn-PI, which is more effective at cross-linking. The C-terminal is found in plasma in two forms of which 70% is the full-length active form (Met-PB-PI) and 30% inactive (Met-NPB-PI). Only the active form can bind to plasmin(ogen) via a two-step process which can either be reversible or irreversible. PI, plasmin inhibitor.
N-Terminal Variation
Native PI contains 464 residues with the first amino acid being methionine.[10]
[12]
[24]
[25] However, only approximately 30% of the circulating PI occurs in this Met–PI form[24] and nearly 70% of the circulating protein resides within an N-terminally truncated form.[26] In this truncated form, the first 12 residues of Met–PI are removed by digesting the peptide bond between P12 and N13 ([Fig. 1]), giving rise to a form of PI with N13 at its extreme N-terminus, a variant called Asn–PI.[26]
[27]
[28] This N-terminal proteolytic processing is performed by a circulating plasma protease named antiplasmin-cleaving enzyme (ACE), which is soluble, C-terminally truncated form of the fibroblast activation protein (FAP), a member of the prolyl oligopeptidase family.[26]
[29]
[30]
[31]
[32]
There is a functional significance and physiological implications of such proteolytic processing: recombinant Met–PI exhibits 66% reduced cross-linking by factor XIII (FXIII) compared with recombinant Asn–PI, while plasma-purified Asn–PI is 13 times faster at cross-linking than plasma-purified Met–PI.[25]
[26] In Met–PI, the major site of fibrin cross-linking occurs via a glutamate residue (E14) at N-terminus[33] which interacts with a lysine residue (K303) on fibrin[34] ([Fig. 1]). However, recent evidence suggests the presence of other cross-linking sites, and this is an area of much-needed study.[35]
Plasma clot stability is reduced in PI-deficient patients.[36]
[37]
[38] The higher activity of Asn–PI variant of the protein is demonstrated by compromised cross-linking into fibrin when the N-terminus is extended by 3 amino acids using recombinant DNA technology,[39] explaining the relationship between fibrinolytic efficiency and Asn–PI plasma levels.[26]
[40]
[41] Taken together, the first 12 amino acids interfere with the cross-linking and reduce the antifibrinolytic effect of PI.[24]
The superior antifibrinolytic effect of Asn–PI explains the functional heterogeneity of PI in individuals with different genetic variants. For example, the conversion of Met–PI to Asn–PI seems to be affected by the presence of a polymorphism of arginine to tryptophan (pR6W, rs2070863) in the SERPINF2 gene, with the sFAP enzyme cleaving the Met–PI(R6) approximately eightfold faster than Met–PI (W6).[42] Human studies indicate that homozygous Met–PI (W6) patients have the shortest clot lysis times.[42] A limited number of population-based studies have indicated a protective effect of W6 allele against ischemic stroke and a minor protective effect against abdominal aortic aneurysm (AAA).[43] However, no such protection was observed against coronary artery disease (CAD) and myocardial infarction (MI). In a recent study on the potential association of the polymorphism (pR6W, rs2070863) with CAD and MI, Bronić and colleagues reported that patients with R6W PI CC genotype had 3.86 times higher odds ratio of risk factor for the CAD than the patients with R6W PI TT genotype in a group of Croatian patients.[44]
C-Terminal Variation
The C-terminal region of PI is highly conserved between human, bovine, and murine species and extended by approximately 55 amino acid residues compared with other serpins.[13]
[45] This region, which contains six lysine residues and mediates electrostatic interactions with the lysine-binding sites (LBS) in plasminogen, is also proteolytically processed.[24]
[46] Also, it contains an arginine-glycine-aspartic acid (RGD) sequence important in cell recognition and integrin adhesion.[47] A synthetic RGD sequence coupled to PI carboxy-terminal sequence inhibited platelet activation and increased plasmin generation, consequently facilitating in vitro fibrin-clot lysis[48]
[49] ([Fig. 1]).
Purification of human and rat PI from plasma has yielded two species with only one having the ability to interact with LBS.[18]
[50]
[51] The plasminogen LBS-binding form synthesized in the liver was termed PB–PI, and the other nonbinding form, formed in the circulation, as NPB–PI.[50]
[52]
[53]
[54] It was found that PB–PI, which accounted for approximately 65% of circulating PI, was the rapid-acting PI, while NPB–PI, which accounted for the remaining 35%, reacted slowly.[24]
[50]
[52]
[53]
[54]
[55]
[56]
[57] It has been demonstrated in vitro that purified PB–PI can spontaneously convert into NPB–PI.[58] This in vitro conversion can also be performed by proteases like trypsin, elastase, and MMP-3, producing a 26-amino-acid C-terminal fragment unique to PB–PI, but the protease responsible for the in vivo conversion is not known.[24]
[59]
[60]
[61]
[62]
[63]
[64] The cleavage also appears to occur at a slightly different site in vivo because monoclonal antibodies raised against the C-terminal region reacted differently with the in vivo- and in vitro-generated NPB–PI.[65] Studies into the importance of the C-terminal lysine residues in inhibiting plasmin/plasminogen have yielded inconsistent results.[24] However, the most conserved C-terminal lysines, K427, K434, K441, K448, and K464, are involved in plasmin binding; residue K464 is particularly important as a K464A mutation causes the greatest reduction in plasmin inhibition.[66]
The C-terminus of PI largely regulates protein activity and its proteolytic removal may be of important clinical consequences.[24] The diagnostic test, antiplasmin activity assay, was found to be dependent on PB–PI.[24] The fraction of circulating PB-PI seems to vary between individuals, ranging from 10 to 60%, but the highest PI activity was obtained from individuals with more PB–PI.[67] Moreover, it appears that the C-terminal variation may also be as important as the N-terminal variation for cross-linking to fibrin because it is PB–PI that is primarily involved in cross-linking.[68] Additionally, a polymorphism in the C-terminus of PI (R407K, rs1057335) which exhibited linkage disequilibrium (LD) with the R6W polymorphism was associated with a significant 23% risk reduction for the development of AAA.[43] Whether this finding is related to a function effect of R407K or simply secondary to LD with R6W is unclear.
Structural Information
To date, structural information for PI has been limited to the murine species, Mus musculus.[14] Valid homology models of the human form of PI can be produced using this structure, given the high sequence identity (75% overall), with 366 identical positions and 83 similar positions. The PI crystal structure consists of residues 46 to 367 and 377 to 419 ([Fig. 3A]). Unfortunately, residues 368 to 376 in the reactive center loop and residues 420 to 464 of the C-terminus could not be fitted into the electron density map, making a complete understanding of how PI may initially interact with plasmin more difficult to comprehend.
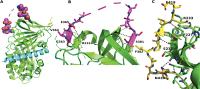 Fig. 3 Crystal structure of murine PI. (A) Overall topology of murine PI with the N terminus shown in cyan, reactive center loop in magenta, and C terminus in yellow. (B) Close up of the reactive center loop (magenta) showing the N-terminus portion (363–365) making several hydrogen bonding interactions (black dashed lines) with residues 214–216 of the main body of PI (green, selected residues labeled). The C-terminal portion (residues 380–382) makes a specific hydrogen bonding interaction with residue K306 on the first strand of the C-sheet. (C) Close up of the contacts between the C-terminal region (yellow) and the main body of PI (green). Several hydrogen bonds are made between the two parts of the protein (black dashed lines); selected residues are labeled. Source: Law et al.
[14]
Fig. 3 Crystal structure of murine PI. (A) Overall topology of murine PI with the N terminus shown in cyan, reactive center loop in magenta, and C terminus in yellow. (B) Close up of the reactive center loop (magenta) showing the N-terminus portion (363–365) making several hydrogen bonding interactions (black dashed lines) with residues 214–216 of the main body of PI (green, selected residues labeled). The C-terminal portion (residues 380–382) makes a specific hydrogen bonding interaction with residue K306 on the first strand of the C-sheet. (C) Close up of the contacts between the C-terminal region (yellow) and the main body of PI (green). Several hydrogen bonds are made between the two parts of the protein (black dashed lines); selected residues are labeled. Source: Law et al.
[14]
Murine PI is a potent inhibitor of human plasmin (ka
4.9010[6] M−1/second−1 and SI 1.02) showing that it is functional and correctly folded, adopting the native serpin fold. The 20 amino acid reactive center loop (residues 363–382) is slightly shorter than most inhibitory serpins (24 residues in antithrombin) and is fully expelled from the A β-sheet, with the N-terminal portion of the loop being tightly packed against the main body of the protein ([Fig. 3B]). Structural comparisons with the antitrypsin/trypsin complex[69] suggest that the reactive center loop may be too short to make significant interactions with plasmin outside of its active site.
The C-terminal domain of PI interacts with the Kringle domain of plasmin to facilitate the formation of the PI/plasmin complex. Tight interactions with residues 410 to 419 and the main body of the protein ([Fig. 3C]) position this sequence less than 30 Å from the reactive center loop, putting it in a suitable position to act as a hook and accelerating the interaction with plasmin. Residues 420 to 464 of the C-terminus, including the known plasmin-binding residue K464, cannot be modeled to the observed electron density, demonstrating that this region is flexible in the absence of plasmin.
Physiological Role(s) of Plasmin Inhibitor
Physiological Role(s) of Plasmin Inhibitor
Circulating free plasmin uses LBS, located in the Kringle domain, to bind PI. However, the LBS is also used by plasmin (and plasminogen) to bind to fibrin.[70] Since LBS on plasmin is shared by both PI and fibrin, they are mutually exclusive: fibrin-bound plasmin cannot bind to PI anymore.[70] This makes plasmin, when bound to fibrin, resistant to inhibition by PI.[70] Conversely, when PI is cross-linked to the fibrin network, it reduces fibrin degradation through inhibition of both plasmin binding and protein activity. PI is cross-linked to fibrin by activated XIII (FXIIIa) and inhibits plasminogen binding to fibrin in direct proportion to the amount incorporated.[36]
[71]
[72] In addition, PI was found to interact with plasminogen binding to fibrin to form a weakly bound complex at a specific site of the plasminogen molecule and the site may be 6-aminohexanoic acid-binding site, resulting in an efficient inhibitor of fibrinolysis.[73]
Binding of plasminogen to fibrin induces a conformational change in plasminogen that is key for activation by tPA.[74] Additionally, the binding of plasminogen to fibrin brings it in close proximity to fibrin-bound tPA, which can then activate and convert plasminogen to plasmin on the fibrin surface.[70] Plasmin starts clot lysis and during this process, more lysine residues are exposed to fibrin, resulting in additional plasminogen/plasmin binding and leading to amplification of the fibrinolysis.[75] This coactivator-mediated sequential activation process ensures the control of clot formation and avoids unregulated thrombus extension.[70]
Plasmin Inhibitor, Diabetes, and Vascular Disease
Plasmin Inhibitor, Diabetes, and Vascular Disease
As alluded to earlier, hypofibrinolysis is a key abnormality in diabetes, historically attributed to elevated levels of PAI-1.[76] However, several other mechanisms were subsequently identified, related to alterations in fibrin network structure, the fibrinolytic system, and incorporation of antifibrinolytic proteins into the clot.[7] Evidence suggests that FXIIIa-catalyzed cross-linking of PI into the fibrin network is enhanced in those with type 1 or type 2 diabetes ([Table 1]). Therefore, this may be one diabetes-specific target for the reduction of the hypofibrinolytic environment in this condition. An earlier study by Fattah and colleagues showed elevated plasma levels of PI in individuals with diabetes.[77] The increased incorporation of PI into fibrin has also been shown in patients with diabetes following MI or ischemic stroke.[78]
Table 1
Summary of different studies investigated the role of PI in patients with diabetes
|
Study
|
Patients
|
Controls
|
Laboratory technique
|
Main findings
|
|
Fibrinolysis parameters
|
Plasma level
|
PI incorporation
|
|
Fattah et al 2003[77]
|
45 individuals with diabetes
Group I: 15 T1D patients
Group II: 15 nonobese (BMI <30 kg/m2), T2D patients
Group III: 15 obese (BMI > 30 kg/m2), T2D patients
|
45 age, sex-matched healthy
|
Not fully described
|
|
Diabetes patients exhibited a significant elevation in PI
T1D vs. control= 143.2 ± 6.5 vs. 103.1 ± 4.5%, p = 0.001
Nonobese T2D vs. control =126.8 ± 5.5 vs. 109.2 ± 4.5%, p = 0.01
Obese T2D vs. control =134.3 ± 5.6 vs. 108.0 ± 4.8%, p = 0.01
|
|
|
Dunn et al 2006[81]
|
150 patients with T2D
|
50 matched controls
|
Confocal microscopy
microtiter plate assay
|
Lysis velocity in T2D vs. control =1.35 ± 0.37 vs. 2.92 ± 0.57 μm/min, p < 0.0001
|
|
PI cross-linking is correlated with HbA1c (r = 0.59, p = 0.001)
|
|
Agren et al 2014[85]
|
236 patients with T1D
|
78 age and sex-matched controls
|
Classical two-site ELISA
turbidimetric analysis
|
Clot lysis time of T1D vs. control = 858 ± 228 vs. 927 ± 208 s, p = 0.009
|
PI levels in plasma of T1D vs. control = 78.5 ± 13.3 vs. 83.2 ± 15.4 mg/L, p = 0.032
|
PI levels in fibrin of T1D vs. control = 1.65 ± 0.25 vs. 1.35 ± 0.18 mg/L, p < 0.001
|
|
Polat et al 2014[84]
|
52 patients with T1D
Group I: 21 without retinopathy
Group II: 18 with non- proliferative retinopathy
Group III: 13 with proliferative retinopathy
|
40 healthy subjects
|
Enzymatic methods
|
|
PI levels of group I = 245 (31–650)
group II = 202 (55–904)
group III = 418 (42–1184)
control = 115.5 (23–591)
p < 0.01, <0.05, <0.001 compared group I,II, and III with control, respectively
|
|
|
Uitte de Willige et al 2018[78]
|
59 arterial thrombotic patients with T2D
|
59 age- and sex-matched arterial patients without T2D, matched for type of arterial thrombosis (myocardial infarction or ischemic stroke)
|
Monoclonal antibody against PI (MA-AP15D7)
|
|
|
PI incorporation in T2D vs. non-T2D = 33.3 ± 4.9% vs. 31.4 ± 5.2%, p = 0.047
|
|
Bryk et al 2020[86]
|
113 T2DM patients (54 women and 59 men)
|
N/A
|
In-house ELISA
Turbidimetric analysis
|
PI incorporation is associated with impairment of fibrinolysis in female patients with T2DM
along with 8.4% longer time to 50% lysis (Lys50MA, p = 0.012)
|
Slightly higher plasma α2-antiplasmin concentration compared with men
(p = 0.005)
|
Female patients had 15.2% greater α2-antiplasmin incorporation into the fibrin clot compared with men
(p = 0.008)
|
Abbreviations: BMI, body mass index; ELISA, enzyme-linked immunosorbent assay; HbA1c, hemoglobin A1c; PI, plasmin Inhibitor; T1D, type one diabetes; T2D, type two diabetes.
The exact mechanism for increased incorporation of PI into diabetes clots is not entirely clear but may be related, at least in part, to the known denser fibrin networks in diabetes,[79]
[80]
[81]
[82] resulting in increased incorporation of the protein. Alternatively, PTMs of PI in diabetes, such as glycation, may increase incorporation into fibrin networks. Bryk and colleagues have recently demonstrated that PI function can be altered following protein glycation.[83] However, only in vitro work has been conducted and it is unclear at present whether PI from diabetes patients undergoes PTMs that affect protein function, and this remains an area for future research.
Moreover, a higher level of PI was associated with more advanced retinopathy and there was a positive correlation between PI and HbA1c levels.[84] Dunn et al showed a relationship between PI incorporation into clots and HbA1c levels in 150 patients with type 2 diabetes.[81] Agren et al found increased incorporation of PI into the fibrin network in patients with type 1 diabetes, although these patients exhibited a contradictory reduction in clot lysis time, which the authors attributed to reduced PAI-1 activity.[85] A more recent study on 113 T2D individuals suggests a gender difference in PI incorporation into fibrin networks with a significant increase in female patients compared with males, which may have clinical implications.[86]
While the number of studies is relatively limited, they collectively suggest that PI plays a role in diabetes complications. However, further research in this area is required to understand the exact subgroups of diabetes patients in whom PI modulates clinical outcomes.
Deficiency of Plasmin Inhibitor
Deficiency of Plasmin Inhibitor
The deficiency of PI is suspected when common conditions for a bleeding disorder are ruled out.[87]
[88] Type I PI deficiency exhibits reduced PI protein levels and activity, while type II deficiency exhibits only reduced activity.[89]
The first congenital deficiency of PI was reported in 1969 in a Japanese patient who was a 16-year-old boy suffering from repeated bleeding and hemorrhage into joints[90] and who had a total absence of the protein. Such congenital PI defects are rare but can cause uncontrolled fibrinolysis and clinically significant bleeding.[91] Heterozygous individuals may be asymptomatic or exhibit milder bleeding symptoms during trauma, dental, or surgical procedures.[65]
[87]
[92]
In contrast, the rare genetic defects acquired deficiency of PI is more common, such as advanced liver and kidney disease and disseminated intravascular coagulation.[93]
[94]
[95]
These findings indicate that complete deficiency (or full suppression) of PI increases bleeding complications, but partial deficiency (or partial suppression) is relatively well-tolerated, and therefore, PI can be considered as a therapeutic target, which is further discussed below.
Targeted Plasmin Inhibitor Modification as Thrombolytic Therapy
Targeted Plasmin Inhibitor Modification as Thrombolytic Therapy
PI presents a potential attractive therapeutic target: inactivation of PI was found to increase the rate of endogenous fibrinolysis to the same extent as that can be attained by high doses of recombinant tPA (r-tPA), but without complications such as the increased risk of surgical bleeding.[96] Moreover, while the genetic deficiency of FX or prothrombin is lethal, the genetic deficiency of PI is well tolerated. Mice with genetic deficiency of PI induced by gene targeting have normal fertility, viability, and coagulation.[97]
Limitations of Current Thrombolytic Therapies
Current drugs administered for thrombolytic therapy, streptokinase (which binds to the catalytic domain of human plasmin),[98] r-tPA (which cleaves the zymogen plasminogen at its Arg561–Val562 peptide bond to form plasmin),[98]
[99] and urokinase plasminogen activator uPA certainly help in clearing the thrombotic blockade in a blood vessel but can also result in unwanted side effects.[100]
[101]
[102]
[103] Even therapy with r-tPA, which is fibrin specific, showed only a marginal improvement in survival.[104]
[105]
[106] Importantly, r-tPA caused significant intracranial bleeding complications that further limits its use. Other serious side-effects of r-tPA include the permeabilization of the blood–brain barrier, cerebral edema, and neurotoxicity. For this reason, r-tPA is only used within the first 4.5 hours of vessel occlusion for stroke treatment, when the benefit/risk ratio is highest,[70]
[101]
[102] while it has been largely superseded by coronary angioplasty for the management of acute myocardial infarction.[107]
Additionally, while r-tPA is effective in lysing fresh clots, it is less efficient against clots aged 4 hours or more, which further limits its practical use[108].Yet another problem with tPA is that it directly generates plasmin from plasminogen, but plasmin can also act as an anticoagulant, degrading clotting factors V, VIII, IX, X, XIIIa, and tissue factor pathway inhibitor.[109] So, for safe thrombolytic therapy, and to prevent a hemophilia-like state, the amount of plasmin produced should be small, but this becomes very difficult to control due to the high therapeutic dose requirement of r-tPA.[70]
[102] In summary, while tPA is effective for acute thrombotic vascular occlusion, its effects are too broad, which translates clinically into a significant increase in bleeding risk. Also, such therapy cannot be used long term, and therefore, alternative fibrinolytic agents are required.
PI inhibition may be particularly valuable in conditions associated with increased protein incorporation into the clot, such as diabetes, which is characterized by a hypofibrinolytic environment that contributes to increased thrombosis risk.
Critical Areas of Plasmin Inhibitor Targeted Drug Development
PI modulates fibrinolysis in three different ways: directly binding to, and rapidly forming a covalent inhibitory complex with plasminogen or plasmin (PAP complex), cross-linking into the fibrin network mediated by FXIIIa making the clot more resistant to plasmin, and inhibition of plasminogen adsorption onto fibrin ([Fig. 4]).[11]
[46]
[110]
[111] Therefore, attempts to inhibit PI function largely rely on these mechanisms as discussed below.
 Fig. 4 Critical areas of PI targeted and therapeutic strategies. (A-C) PI modulates fibrinolysis in different ways: directly binding to, and rapidly forming a covalent inhibitory complex with plasminogen or plasmin (PAP complex), cross-linking into the fibrin network, mediated by FXIIIa, and inhibiting plasminogen adsorption onto fibrin. As such, attempts to develop any drug inhibiting PI (D), tackle one of these areas (1) PI mimicking peptides, (2) inhibitors of ACE, (3) monoclonal antibodies inhibiting PI, and (4) microplasmin. ACE, antiplasmin-cleaving enzyme.
Fig. 4 Critical areas of PI targeted and therapeutic strategies. (A-C) PI modulates fibrinolysis in different ways: directly binding to, and rapidly forming a covalent inhibitory complex with plasminogen or plasmin (PAP complex), cross-linking into the fibrin network, mediated by FXIIIa, and inhibiting plasminogen adsorption onto fibrin. As such, attempts to develop any drug inhibiting PI (D), tackle one of these areas (1) PI mimicking peptides, (2) inhibitors of ACE, (3) monoclonal antibodies inhibiting PI, and (4) microplasmin. ACE, antiplasmin-cleaving enzyme.
Decoy Protein Incorporation
Having a decoy protein competing with PI in cross-linking to fibrin reduces the amount of PI incorporated into the clot, thus enhancing fibrinolysis. Kimura and colleagues produced a N-terminal 12 residue PI-based peptide that reduced PI cross-linking into the fibrin clot, thus facilitating lysis.[112] Two decades later, similar work on another decoy protein showed only a partial effect[113].
Sheffield and colleagues introduced a more sophisticated decoy protein using DNA manipulation technology to produce the chimeric protein of N-terminal PI (13–42) with human serum albumin (HSA).[114] Chimeric PI–HSA competed with PI cross-linking into clot and accelerated clot lysis. However, the chimeric protein was a less effective substrate for cross-linking into fibrin networks than PI, requiring an excessive amount (14-fold molar excess).[114] None of these “decoy proteins” advanced into human studies for reasons that are not entirely clear.
Given the established functional sites of PI, one group mutated residue R364 to alanine in the active site of PI. This modified version of the protein functioned as a FXIIIa substrate with affinity and kinetic efficiency comparable to those of native PI, despite having an additional acetylated Met blocking group at its amino-termini. It was cross-linked into fibrin networks but partly lost the ability to inhibit plasmin, in turn enhancing fibrinolysis.[71]
[115] This indicates that some residues on PI (such as R364) are not affected by FXIII cross-linking but partly lose PI activity, thus representing potential therapeutic targets.
Microplasmin
Microplasmin is a cleaved version of plasmin that contains only the catalytic domain, and it is mainly inhibited by α2-macroglobulin and by PI.[116] Its enzymatic activity is similar to that of plasmin.[117] Unlike tPA, microplasmin is a direct-acting thrombolytic, as compared with most other thrombolytics which dissolve clots indirectly by activating the plasmin precursor, plasminogen. Being a truncated derivative of plasmin lacking the LBS, it has no affinity for fibrin and it is inhibited more slowly by PI than intact plasmin.[116]
[118] Infusion of microplasmin into mouse and hamster models depleted circulating PI thus reducing experimental cerebral ischemic injury and improving neurological outcome.[119]
[120]
[121] A phase I human study of intravenous microplasmin showed a dose-dependent decrease in PI activity in healthy volunteers.[122] In a phase II trial in stroke subjects, microplasmin decreased systemic PI levels up to 80% but had no effect on hard clinical outcomes.[123] Of note, microplasmin administration was associated with reduced fibrinogen levels, indicating limited specificity for fibrin and questioning its use in clinical practice.
Reduction in Plasmin Inhibitor Cleavage
Researchers attempted to inhibit the conversion of Met–PI to Asn–PI, the more powerful antifibrinolytic version of the protein.[26] A substrate analog inhibitor, Phe-Arg-(8-amino-3,6-dioxaoctanoic acid)-Gly-[r]-fluoropyrrolidine, inhibited APCE with a Ki
of 54 μM but no dipeptidyl peptidase IV even at 2 mM. The inhibitor also blocked the cleavage of Met–PI with an IC50 of 91 μM. High-affinity inhibitor of ACE demonstrated a decrease in the proteolytic conversion of Met–PI to Asn–PI in a concentration-dependent manner, with shortened lysis time of plasma clots.[41]
[124] However, in vivo studies are lacking and the clinical applicability of this approach is uncertain. Circulating ACE is structurally similar to FAP. Molecular modeling showed that the active site of FAP has a large central pore that can accommodate the N-terminal region of Met–PI, explaining the specificity of ACE to this form/structure of the protein.
Direct Modulation of Plasmin Inhibitor
Monoclonal antibodies against PI have been developed to modulate protein function and showed promise in different animal models of venous thrombosis, pulmonary embolism (PE), and ischemic stroke.[125]
[126]
[127]
[128]
[129]
[130]
[131]
[132]
[133]
[134]
[135]
[136]
[137] A monoclonal antibody against PI that interferes with the formation of PAP complexes has been shown to increase the effectiveness of tPA-mediated clot lysis.[133] Another monoclonal antibody against PI has demonstrated synergistic interactions with plasminogen activators and enhanced fibrinolysis by increasing the potency of streptokinase, t-PA, and urokinase (by 20, 27, and 80-fold, respectively).[131]
In addition, Kumada and Abiko reported a reduction in the level of circulating PI levels in rats by repeated injection of polyclonal anti-PI F(ab')2 fragments and which accelerated thrombolysis.[138] A rabbit jugular vein thrombosis model suggested that a combination of a PI inhibitor and a plasminogen activator may be a more potent thrombolytic strategy.[36]
Similarly, in a humanized model of acute PE in mice, Singh et al[125] found that thrombus that targeted PI by TS23 (a monoclonal antibody neutralizing PI activity) enhanced the dissolution of pulmonary emboli, without an increased bleeding risk.[125] Anti-PI TS23 also blocked thrombus formation during venous stasis in mice,[132] and this agent was tested in phase I trials in healthy volunteers (NCT03001544) but is yet to be tested in pathological states.[139]
The monoclonal PI antibody DS-9231 has been tested in clinical phase I/II study in individuals with PE, but the compound was subsequently withdrawn for reasons that are yet to be announced (ClinicalTrials.gov; NCT03316729). While these monoclonal antibodies have shown inhibition of PI function, the mode of action is unknown given the absence of data on interaction sites and it remains unclear whether they result in changes to the protein structure.
Conclusions and Future Directions
Conclusions and Future Directions
Hypofibrinolysis is associated with vascular disease and is directly linked to adverse clinical outcome following an ischemic event. PI is a strong inhibitor of fibrinolysis and therefore represents a credible therapeutic target to reduce thrombosis risk. Moreover, targeting PI may be particularly beneficial in conditions with increased protein incorporation into the clot, such as diabetes, thus offering a disease-specific therapy. Various strategies have been used to inhibit PI function including the use of decoy protein, thus reducing PI incorporation into clots, inhibition of Asn–PI production, the stronger version of the protein, and direct modulation of protein activity using monoclonal antibodies. In vitro and animal in vivo studies showed promise but none of the numerous “PI-inhibitors” have made it into routine clinical use. The exact reasons for the failure to convert early success into clinical use are not always clear and may be due to the financial viability of such an approach or safety concerns although no unexpected adverse events have been reported to date.
Despite the failure to translate PI inhibition into clinical use, advances in technology are likely to identify alternative ways of modulating the PI activity or the amount of protein incorporated into the clots. This in turn will offer a novel therapeutic opportunity to reduce thrombosis risk while keeping bleeding risk to a minimum. Future translational research is required to identify effective and safe inhibitors of PI, which will hopefully be used to reduce the risk of both arterial and venous thrombotic events.
Supplementary Material S1 Literature Search
Supplementary Material S1 Literature Search
We searched literatures from MEDLINE® and EMBASE® using terms described as follow: Antiplasmin (or alpha 2 antiplasmin, plasmin inhibitor, Serpin F2) +thrombosis +diabetes; Antiplasmin (or alpha 2 antiplasmin, plasmin inhibitor, Serpin F2) +cardiovascular +diabetes; Antiplasmin (or alpha 2 antiplasmin, plasmin inhibitor, Serpin F2) +coronary artery +diabetes; Antiplasmin (or alpha 2 antiplasmin, plasmin inhibitor, Serpin F2) +stroke +diabetes; Antiplasmin (or alpha 2 antiplasmin, plasmin inhibitor, Serpin F2) + (Inactivation, modulation, inhibition, antibody, nanobody, nanoparticle or small molecule) ; Antiplasmin (or alpha 2 antiplasmin, plasmin inhibitor or Serpin F2) +(fibrinolysis, fibrin clot lysis time, clot).





