Keywords
oligodendroglioma - oligosarcoma - co-deletion 1p/19q - spinal metastasis
Palavras-chave
oligodendroglioma - oligossarcoma - co-deleção 1p/19q - metástases raquidianas
Introduction
Oligodendrogliomas are infiltrative tumors of the central nervous system (CNS). They
constitute between 4 and 5% of the primary CNS tumors and ∼ 4 to 15% of the glial
tumors.[1] Among the glial tumors, oligodendrogliomas have always been considered morphologically
stable.[2]
Gliosarcomas are rare primary tumors of the CNS, histologically composed of glial
and sarcomatous cells, with the glial component being, in most cases, astrocytic.[3]
We describe here a case of an oligosarcoma (World Health Organization [WHO] grade
III[4]), originating from an oligodendroglioma (WHO grade II[4]), in a patient not previously submitted to complementary treatments.
Case Report
A 36-year-old man presented with an onset of convulsive crisis in 2014. He underwent
brain computed tomography (CT) that showed a left frontal intra-axial lesion with
calcifications in the interior ([Fig. 1]) and brain magnetic resonance imaging (MRI) that showed a lesion at the same location
with hyperintensity T2 and without contrast enhancement, suggestive of low-grade glioma
([Fig. 2]). The patient was submitted to craniotomy and gross total resection (GTR) of the
lesion ([Fig. 3]). Histological analysis showed a tumor with round nuclei cells and perinuclear halos,
with extensive areas of calcification. It also showed absence of mitoses, necrosis
and showed low nuclear proliferation index. The lesion was classified as oligoastrocytoma
WHO grade II, according to the WHO tumor classification of 2007[5] ([Figs. 4] and [5]), and the tumor was positive for the presence of 1p/19q co-deletion. The patient
remained under clinical and imaging surveillance, without any complementary treatment.
 Fig. 1 Computed tomography scan showed a left frontal lesion with calcification.
Fig. 1 Computed tomography scan showed a left frontal lesion with calcification.
 Fig. 2 T2-weighted magnetic resonance imaging showed a hyperintensity left frontal lesion.
Fig. 2 T2-weighted magnetic resonance imaging showed a hyperintensity left frontal lesion.
 Fig. 3 First surgery postoperative magnetic resonance imaging showed gross total resection
of the tumor.
Fig. 3 First surgery postoperative magnetic resonance imaging showed gross total resection
of the tumor.
 Fig. 4 HE staining showed round nuclei with a perinuclear halo, and a low proliferation
index.
Fig. 4 HE staining showed round nuclei with a perinuclear halo, and a low proliferation
index.
 Fig. 5 HE staining showed round nuclei with a perinuclear halo, and a low proliferation
index.
Fig. 5 HE staining showed round nuclei with a perinuclear halo, and a low proliferation
index.
Two years later, in imaging control, tumor growth was recorded in the posterior and
medial portion of the surgical site. An MRI showed characteristics similar to the
initial lesion, namely without evidence of contrast enhancement, with spontaneous
hyperintensity in T2-fluid-attenuated inversion recovery (FLAIR) weighted imaging
([Figs. 6] and [7]). The patient underwent a new surgical procedure with GTR of the recurrent tumor
([Fig. 8]). The neuropathological characteristics confirmed that it was an oligodendroglioma
WHO grade II, isocitrate dehydrogenase (IDH-1) positive (according to the WHO classification
of 2016 CNS tumors[4]), maintaining the presence of 1p/19q co-deletion and α thalassemia/mental retardation
syndrome X-linked (ATRX) mutation positive ([Figs. 9] and [10]), now with a moderate nuclear proliferation index. The patient was once again referred
to clinical and imaging surveillance, not considering indications for adjuvant treatments,
given the clinical, imaging, anatomopathological and degree of tumor removal obtained.
 Fig. 6 T2-fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging showed
a tumor recurrence in the posterior and medial walls of the surgical site.
Fig. 6 T2-fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging showed
a tumor recurrence in the posterior and medial walls of the surgical site.
 Fig. 7 T2-fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging showed
a tumor recurrence in the posterior and medial walls of the surgical site.
Fig. 7 T2-fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging showed
a tumor recurrence in the posterior and medial walls of the surgical site.
 Fig. 8 Second surgery postoperative magnetic resonance imaging showed gross total resection
of the tumor.
Fig. 8 Second surgery postoperative magnetic resonance imaging showed gross total resection
of the tumor.
 Fig. 9 Hematoxylin & eosin staining with de same initial type of cells, but with a moderate
proliferation index.
Fig. 9 Hematoxylin & eosin staining with de same initial type of cells, but with a moderate
proliferation index.
 Fig. 10 Hematoxylin & eosin staining with de same initial type of cells, but with a moderate
proliferation index.
Fig. 10 Hematoxylin & eosin staining with de same initial type of cells, but with a moderate
proliferation index.
After 12 months, new MRI control showed tumor recurrence, now with a contrast enhancement
area, suggesting possible dedifferentiation ([Figs. 11] and [12]). It was operated on with a GTR of the lesion, as evidenced by postoperative brain
CT ([Fig. 13]). The histological analysis showed a very cellular tumor, with frequent mitoses,
areas of extensive necrosis, with rounded nuclei cells, and an evident cytoplasm and
a fasciculated aspect. The immunohistochemistry study showed positivity for glial
fibrillary acidic protein (GFAP), and the sarcomatous portion was strongly positive
for vimentin. It maintained positivity for IDH-1 and ATRX and presented a very high
proliferation index. The neuropathological study now showed the occurrence of sarcomatous
transformation, maintaining the oligodendroglial component (oligosarcoma WHO grade
III [4]), ([Figs. 14]
[15]
[16]
[17]
[18]
[19]
[20]). With this new histological diagnosis, the patient started complementary treatment
according to the Stupp protocol. He completed 30 sessions of radiotherapy (2 Gy/session,
totalizing 60 Gy), with concomitant temozolomide 75 mg/m2 for 7 days/week, for 6 weeks. Following this, the patient underwent 1 cycle of adjuvant
temozolomide 150 mg/m2 for 5 days.
 Fig. 11 T1-weighted magnetic resonance imaging showed a left frontal lesion with contrast
enhancement (12).
Fig. 11 T1-weighted magnetic resonance imaging showed a left frontal lesion with contrast
enhancement (12).
 Fig. 12 T1-weighted magnetic resonance imaging showed a left frontal lesion with contrast
enhancement (12).
Fig. 12 T1-weighted magnetic resonance imaging showed a left frontal lesion with contrast
enhancement (12).
 Fig. 13 Third surgery postoperative computed tomography scan without contrast, showed gross
total resection of the tumor.
Fig. 13 Third surgery postoperative computed tomography scan without contrast, showed gross
total resection of the tumor.
 Fig. 14 Hematoxylin & eosin staining showed hypercellular tumor with round nuclei, with a
perinuclear halo, with frequent mitoses and areas of extensive necrosis.
Fig. 14 Hematoxylin & eosin staining showed hypercellular tumor with round nuclei, with a
perinuclear halo, with frequent mitoses and areas of extensive necrosis.
 Fig. 15 Hematoxylin & eosin staining showed hypercellular tumor with round nuclei, with a
perinuclear halo, with frequent mitoses and areas of extensive necrosis.
Fig. 15 Hematoxylin & eosin staining showed hypercellular tumor with round nuclei, with a
perinuclear halo, with frequent mitoses and areas of extensive necrosis.
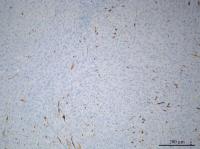 Fig. 16 Immunohistochemistry showed positive staining for glial fibrillary acidic protein
(GFAP).
Fig. 16 Immunohistochemistry showed positive staining for glial fibrillary acidic protein
(GFAP).
 Fig. 17 The tumor showed positive staining for vimentin in the sarcomatous portion.
Fig. 17 The tumor showed positive staining for vimentin in the sarcomatous portion.
 Fig. 18 The tumor was positive for IDH-1.
Fig. 18 The tumor was positive for IDH-1.
 Fig. 19 The tumor was positive for α thalassemia/mental retardation syndrome X-linked (ATRX).
Fig. 19 The tumor was positive for α thalassemia/mental retardation syndrome X-linked (ATRX).
 Fig. 20 The proliferation index was very high.
Fig. 20 The proliferation index was very high.
During the 2nd cycle of chemotherapy, the patient presented with a new neurological
deficit, a motor apraxia of the right upper limb. Another MRI was undertaken, which
revealed tumor progression, with a recurrent tumor in the surgical site and still
with left hemispheric meningeal dissemination revealing an extra-axial mass with contrast
enhancement ([Figs. 21] and [22]). A new surgical intervention with GTR of the various relapsed lesions, as documented
by postoperative MRI ([Figs. 23] and [24]), was performed. The parietal lesion corresponded to a mass of hard consistency
and was extra-axial ([Figs. 25] and [26]). The neuropathological study of the various lesions was shown to be the same sarcomatous
tumor, and complementary treatment with 2nd line chemotherapy (irinotecan with bevacizumab)
was initiated. At the end of 2 months of treatment, the patient exhibited severe cervical
radiculopathy without relief with analgesics, and was submitted to lumbar puncture,
which showed the presence of malignant cells in the cerebrospinal fluid. He underwent
a neuro-axis MRI, which showed stability of the disease at the brain but extensive
spreading to the medulla with intradural and extramedullary lesions at the cervical
and medullary cone levels ([Figs. 27], [28]). He started treatment with 3rd line chemotherapy (lomustina 90 mg/m2 for 6 days/week, for /6 weeks, and bevacizumab 10 mg/kg for 2 days/week for 2 weeks.
The patient also performed 5 sessions of radiotherapy directed to the cervical lesion
(5 fractional radiotherapy sessions totaling 20 Gy). Currently, the patient has a
4-year overall survival, with Karnofsky performance status of 60% and eastern cooperative
oncology group (ECOG) performance status 2.
 Fig. 21 T1-weighted magnetic resonance imaging showed a recurrent tumor in the surgical site
and a left hemispheric extra-axial mass with contrast enhancement (22).
Fig. 21 T1-weighted magnetic resonance imaging showed a recurrent tumor in the surgical site
and a left hemispheric extra-axial mass with contrast enhancement (22).
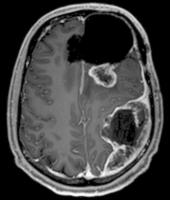 Fig. 22 T1-weighted magnetic resonance imaging showed a recurrent tumor in the surgical site
and a left hemispheric extra-axial mass with contrast enhancement (22).
Fig. 22 T1-weighted magnetic resonance imaging showed a recurrent tumor in the surgical site
and a left hemispheric extra-axial mass with contrast enhancement (22).
 Fig. 23 Fourth postoperative magnetic resonance imaging showed gross total resection of the
tumor.
Fig. 23 Fourth postoperative magnetic resonance imaging showed gross total resection of the
tumor.
 Fig. 24 Fourth postoperative magnetic resonance imaging showed gross total resection of the
tumor.
Fig. 24 Fourth postoperative magnetic resonance imaging showed gross total resection of the
tumor.
 Fig. 25 Intraoperative images showing a lesion of hard consistency, with plane of separation
of adjacent brain parenchyma.
Fig. 25 Intraoperative images showing a lesion of hard consistency, with plane of separation
of adjacent brain parenchyma.
 Fig. 26 Intraoperative images showing a lesion of hard consistency, with plane of separation
of adjacent brain parenchyma.
Fig. 26 Intraoperative images showing a lesion of hard consistency, with plane of separation
of adjacent brain parenchyma.
 Fig. 27 Cervical T1-weighted magnetic resonance imaging with contrast showed a lesion at
C6-C7 level with contrast enhancement.
Fig. 27 Cervical T1-weighted magnetic resonance imaging with contrast showed a lesion at
C6-C7 level with contrast enhancement.
 Fig. 28 Lumbar T1-weighted magnetic resonance imaging with contrast showed a lesion at T12-L1
level with contrast enhancement.
Fig. 28 Lumbar T1-weighted magnetic resonance imaging with contrast showed a lesion at T12-L1
level with contrast enhancement.
Discussion
Isocitrate dehydrogenase-mutated and 1p/19q-positive co-deletion oligodendrogliomas
are considered slow-growing tumors with a better prognosis than the other gliomas.[5] The appearance of sarcomatous tumors at sites of oligodendrogliomas resection in
patients not undergoing further chemotherapy and/or radiotherapy treatments is very
rare.[6] Although in most cases the glial component of the sarcomatous tumors is astrocytic,
the literature describes several cases of gliosarcomas in which the glial component
is oligodendrocytic.[2]
[3]
[6]
[7]
[8]
[9]
[10]
Here, we describe the case of an oligodendroglioma WHO grade II with 1p/19q co-deletion,
IDH-1 and ATRX mutation, with initial GTR, not subjected to complementary treatments,
and which was dedifferentiated to the sarcomatous form. The tumor always maintained
the same genetic characteristics. Despite the presence of predictive factors of better
prognosis, the tumor displayed poor response to radiotherapy and chemotherapy, and
even presented spinal metastasis. Although several cases have been described in the
literature of oligodendrogliomas with transformation to the sarcomatous form (oligosarcoma),
research performed in PubMed and Google Scholar reveals only one case with spinal
metastasis.[8]
Most cases with extracranial dissemination are associated with extensive progression
of the brain tumor;[11] yet, in this case, the spread occurred with stability of the brain lesions.
This case describes a patient with a low-grade glioma, with most of the predictive
factors of better prognosis (age < 40 years, total initial resection and favorable
genetics), in whom progression occurred rapidly and with refractoriness to complementary
treatments.
Conclusion
Despite all the good prognostic factors present in this clinical case and the absence
of previous adjuvant therapies, the tumor was dedifferentiated to a malignant form,
quickly and without any warning signs.
This leads us to conclude that there is a need for further studies that may indicate
new prognostic factors, such as imaging, anatomopathological and genetic characteristics
that help us understand which tumors will dedifferentiate more quickly and which may
respond better to complementary treatments.






