Introduction
π-Conjugated molecules consisting of electron donor (D) and acceptor (A) units have
attracted considerable attention due to their enhanced polarizability, narrow energy
gap, and high carrier mobility, which render them actively used in organic semiconductor
devices.[1] In particular, the D–A type molecules have made significant progress in the exploitation
of organic light-emitting diodes (OLEDs).[2] Nevertheless, most of the π-conjugated luminophores exhibit weak emission behavior
in their aggregated states due to the notorious aggregation-caused emission quenching,
which limits the development of efficient OLED devices.[3] Toward this end, aggregation-induced emission (AIE) has emerged as an effective
strategy to achieve high emission in concentrated solution or even in the solid state.[4] For instance, the carbon-rich tetraphenylethene and its derivatives have been widely
explored to develop high-performance OLEDs.[3]
[4] Despite the progress made in recent years, the bottom-up synthesis of AIE-active
boron–nitrogen (B−N) containing luminogens remains a challenge.[5]
B−N-doped polycyclic aromatic hydrocarbons (PAHs) exhibit broad absorption and intense
fluorescence which can be used as efficient emitters.[6] In addition, the BN-coordinated unit (B←N) and the C−C unit are also recognized
as isoelectronic and isosteric ([Figure 1a]).[7] Unlike the nonpolar covalent C−C bond, the B←N bond possesses a large dipole moment
and a low bond dissociation energy ([Figure 1a]).[8] Accordingly, a B←N unit can serve as a typical kind of Lewis acid/base pair, which
has been utilized to synthesize π-conjugated systems with narrow band gaps, high electron
affinity, and photochromic properties.[7]
[9] For instance, with respect to the unstable phenalene ([Figure 1b]),[10] upon replacing the C−C unit by a B←N unit, the resultant BN-doped phenalene (BNP)
exhibits high ambient stability and intense fluorescence.[11]
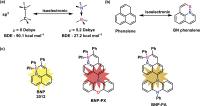 Figure 1 (a) The isoelectronic relationship between B←N coordinated unit and C−C bond. (b)
The isoelectronic relationship between phenalene and B←N-doped phenalene; the substituents
are omitted for clarity. (c) The design principle toward twisted D–A B←N coordinated
phenalene complexes.
Figure 1 (a) The isoelectronic relationship between B←N coordinated unit and C−C bond. (b)
The isoelectronic relationship between phenalene and B←N-doped phenalene; the substituents
are omitted for clarity. (c) The design principle toward twisted D–A B←N coordinated
phenalene complexes.
Herein, we demonstrate the synthesis of two novel D–A phenalene derivatives (BNP-PX and BNP-PA; [Figure 1c]) by employing BNP as the building block. The phenoxazine and phenylphenazine groups in BNP-PX and BNP-PA not only serve as electron donors but also lead to the highly twisted geometrical
conformations, which render the spatial separation of their HOMO and LUMO. Compared
to the parent BNP, the frontier orbital energy gaps of BNP-PX and BNP-PA can be increased by the introduction of the phenoxazine and phenylphenazine substituents.
In addition, BNP-PX demonstrates the largest Stokes shift (8,033 cm−1) among all the reported B←N coordinated complexes,[12] and exhibits excellent AIE behavior with a quantum yield (Φ) of 0.54 in the solid
state. We further fabricated a red OLED device based on BNP-PX, revealing its promises in organic optoelectronic devices.
Results and Discussion
The synthetic routes toward BNP-PX and BNP-PA are depicted in [Scheme 1]. First, the Buchwald–Hartwig coupling of 8-(benzyloxy)-5-bromoquinoline (3) with phenoxazine (1) was performed to afford 10-(8-(benzyloxy)quinolin-5-yl)-10H-phenoxazine (4) in 65% yield. The treatment of 4 with 2 equivalents of 1,4-cyclohexadiene provided 5-(10H-phenoxazin-10-yl)quinolin-8-ol (6) and the crude product was used without purification in the next step. Subsequently,
compound 6 was reacted with trifluoromethanesulfonic anhydride (Tf2O)to afford 5-(10H-phenoxazin-10-yl)quinolin-8-yl trifluoromethanesulfonate (8) in 97% yield over two steps ([Scheme 1]).[13] Next, Pd-induced cyclization of 8 in toluene at 60 °C in the presence of alkynyl(triaryl)borates provided the target
compound 10-(1,1,2-triphenyl-1H-1λ4,10λ4-[1,2]azaborinino-[5,6,1-ij]quino-lin-6-yl)-10H-phenoxazine (BNP-PX) in 91% yield. Following a similar synthetic strategy, (E)-5-(8-(2-(diphenylboraneyl)-2-phenylvinyl)quinolin-5-yl)-10-phenyl-5,10-dihydrophenazine
(BNP-PA), in which the substituent phenoxazine in BNP-PX was replaced by a phenylphenazine unit, was successfully synthesized starting from
compound 3 over four steps ([Scheme 1]).[14] The targeted compound BNP-PX was purified by silica column chromatography and then recrystallized in chloroform/methanol
(CHCl3/MeOH), while BNP-PA was obtained by precipitation in MeOH followed byrecrystallization in CHCl3/MeOH. Compounds BNP-PX and BNP-PA were fully characterized by high-resolution mass spectroscopy (HRMS; [Figures S39], [S40]) and 1H, 13C, 11B as well as 2D NMR measurements ([Figures S28]
[S29]
[S30]
[S31]
[S32]
[S33]
[S34]
[S35]
[S36]
[S37]
[S38]).
 Scheme 1 Synthetic routes toward compounds BNP-PX and BNP-PA.
Scheme 1 Synthetic routes toward compounds BNP-PX and BNP-PA.
Single crystals of compounds BNP-PX and BNP-PA were obtained by slow diffusion of a chloroform/methanol mixed solution. BNP-PX crystallizes in the monoclinic space group P21/c and shows an edge-on twisted geometry ([Figure 2a, b]). Due to the different rotation angles between the BNP and phenoxazine group (R1),
two different conformational isomers (BNP-PXa and BNP-PXb in [Figure 2a, b]) are found in the solid state of BNP-PX. The bond length of B1–N1 (1.642 Å in BNP-PXa and 1.643 Å in BNP-PXb;[Figure 2a, b]) is a typical B←N coordination bond (1.6–1.7 Å),[5a]
[15] which is similar to that of reported BNP ([Figure S1]).[11] Besides, the dihedral angles of C11–N1–B1–C10 in BNP-PXa and BNP-PXb are 7.3 ° and 1.7 ° ([Figure 2a, b]), respectively, indicating the nearly planar geometry of the BNP core. Interestingly,
the dihedral angles between the BNP and phenoxazine group (C4–C5–N2–C30) in BNP-PXa and BNP-PXb are 76.6 ° and 68.0 ° ([Figure 2a, b]), respectively. As shown in [Figure 2d], BNP-PX displays a slipped stack mode with a π–π stacking distance of 3.17 Å (BNP-PXa) and 3.45 Å (BNP-PXb), respectively, which are shorter than the sum of the van der Waals radii (3.60 Å).[16] Similar to BNP-PX, BNP-PA also stacks in themonoclinic space group P21/c and adopts a twisted geometry ([Figure 2c]). Although the size of the phenylphenazine group (R2) is comparable to that of the
phenoxazine group (R1), only one conformation ([Figure 2c]) was found in the single-crystal structure of BNP-PA. This is possibly due to the existence of an additional phenyl unit in R2, which
increases the rotational energy between the R2 and BNP. The bond length of B1–N1 (1.638 Å)
in BNP-PA is slightly shorter than those of BNP and BNP-PX. Moreover, the dihedral angle of C7–C6–N2–C41 in BNP-PA is 70.5 ° ([Figure 2c]), and there is no obvious π–π interaction in the stacking mode of BNP-PA ([Figure 2e]).
 Figure 2 (a, b) Top and side views of single-crystal structure of BNP-PX (isomer BNP-PXa and BNP-PXb). (c) Top and side views of single-crystal structure of BNP-PA. (d) Crystal packing of BNP-PX,blue: isomer BNP-PXa, green: isomer BNP-PXb. (e) Crystal packing of BNP-PA. 50% probability of thermal ellipsoids; colors: black, carbon; blue, nitrogen; pink,
boron; R1: phenoxazine group; R2: phenylphenazine group. Hydrogen atoms are omitted
for clarity.
Figure 2 (a, b) Top and side views of single-crystal structure of BNP-PX (isomer BNP-PXa and BNP-PXb). (c) Top and side views of single-crystal structure of BNP-PA. (d) Crystal packing of BNP-PX,blue: isomer BNP-PXa, green: isomer BNP-PXb. (e) Crystal packing of BNP-PA. 50% probability of thermal ellipsoids; colors: black, carbon; blue, nitrogen; pink,
boron; R1: phenoxazine group; R2: phenylphenazine group. Hydrogen atoms are omitted
for clarity.
The UV-vis absorption and fluorescence spectra of BNP-PX and BNP-PA in anhydrous toluene solution are presented in [Figure 3a]. BNP-PX displays a maximum absorption peak at 424 nm, with a bathochromic shift of 7 nm compared
with BNP ([Figure S52] and [Table S3]). Based on the time-dependent density functional theory (TD-DFT) calculations, this
absorption peak of BNP-PX can be attributed to the HOMO-1→LUMO transition ([Figure S60]) and belongs to the local excitation (LE) from the core of BNP according to the
corresponding orbital plots ([Figure S57]). In addition, a weak shoulder absorption at a longer wavelength of BNP-PX (450 nm; [Figure 3b]) was also observed, which is owing to the intramolecular charge transfer (ICT) from
the donor phenoxazine group to the acceptor BNP on the basis of measured negative
solvatochromic effect[5a]
[17] ([Figure S41] and [Table S2]) and TD-DFT calculations (the HOMO→LUMO transition; [Figures S60] and [S57]). The blue shift of the absorption peak with the increased polarity of the solvent
([Figure S41]) suggests that the ground state of BNP-PX is more polar than its excited state, which is consistent with the molecular donor–acceptor
(D–A) structure.[5a]
[17] For compound BNP-PA, a similar absorption peak at λ
max = 424 nm was observed. According to the TD-DFT calculations, this peak can be assigned
to the HOMO-1→LUMO transition ([Figure 3a]) and also belongs to the LE from the BNP. In addition, there is a broad absorption
from 500 to 700 nm for BNP-PA (maximum peak at 581 nm; [Figure 3b]), because of the ICT effect from the donor phenylphenazine group to the acceptor
BNP based on the negative solvatochromic effect[17] investigation ([Figure S42]) and TD-DFT calculations (the HOMO→LUMO transition; [Figures S61] and [S58]). BNP-PA displays a stronger HOMO–LUMO absorption than BNP-PX ([Figure 3a, b]), this is due to the strong electron-donating character of the sp3-hybridized phenylphenazine group than that of the phenoxazine unit. Moreover, the
weak HOMO–LUMO absorptions of BNP-PA and BNP-PX are also observed in different solvents ([Figures S41] and [S42]), which are well supported by the TD-DFT calculations. The fluorescence spectra
of BNP-PX show a maximum emission peak at 643 nm (excited at 424 nm) with a Stokes shift of
8,033 cm−1 in toluene ([Figure 3c]), exhibiting the largest Stokes shift among all the reported B–N-coordinated complexes.[12] However, two maximum peaks (around 460 and 525 nm) with an additional shoulder (around
675 nm) in the emission spectra of BNP-PX in dichloromethane (DCM), THF, and DMF were observed ([Figure S43]), this is likely attributable to the existence of the twisted ICT.[18] For compound BNP-PA, the maximum emission peak at 518 nm can be observed in toluene (excited at 424 nm).
Moreover, BNP-PA demonstrated a positive solvatochromism of the emission spectra ([Figure S44]), which may due to the fact that the interaction of BNP-PA with different polar solvents is mostly stabilized by the local excited states (as
predicted by the TD-DFT calculations).[19] In addition, no emission spectra of BNP-PA was observed after excitation at ca. 600 nm in different solvents ([Figure S45]), indicating that the emission spectra of BNP-PA ([Figure 3c]) mainly came from the second excited state (the HOMO-1→LUMO excitation, which was
predicted by TD-DFT calculations). The solution quantum yield (QY) of BNP is estimated to be 0.93 (fluorescein as reference; [Table S3]). In contrast, the solution QY of BNP-PX and BNP-PA is estimated to be 0.03 and 0.01 in toluene (Rhodamine 101 and fluorescein as reference;
[Table S3]), respectively. While the solid-state QY of BNP-PX is 0.54 based on the measurement by an integral sphere. Such low solution QY of BNP-PX is possibly due to the free intramolecular motions between the BNP and phenoxazine
group in dilute solvents.[20] Furthermore, the fluorescence behavior of BNP-PX in THF–water solvents with different water volume fractions (f
w) was recorded. As shown in [Figure 3 (e, f)], the fluorescence spectra of BNP-PX displayed an almost flat line in pure THF solution. Interestingly, when the water
fraction was more than 40% (f
w), the fluorescence intensity of BNP-PX increased sharply and reached to the highest intensity at 60% (f
w) ([Figure 3f, g]). Afterwards, it gradually decreased upon increasing the fractions of water. These
results strongly suggest a typical solvent-dependent AIE character for BNP-PX. In contrast, there was no AIE response for BNP and BNP-PA under the same conditions ([Figures 3f], [S50], and [S51]). According to the concentration-dependent absorption and fluorescence spectra ([Figure S47]), there is no obvious excimerformation of BNP-PX under high concentrations. According to the temperature-dependent absorption and
fluorescence spectra ([Figure S48]), the intensity of the HOMO–LUMO absorption and the maximum emission peak of BNP-PX increased with temperature. Thus, we consider that the unique AIE response for BNP-PX might be caused by a restriction of intramolecular motion mechanism,[20] where the free rotational motion of the phenoxazine group in BNP-PX is limited in the aggregated state, resulting in the cut off of nonradiative transition
and intense luminescence.[21] In addition, BNP-PX showed similar solvent-dependent AIE behavior in the THF–hexane solvent ([Figure S49]).
 Figure 3 (a) UV-vis absorption spectra of BNP-PX and BNP-PA in toluene (concentration: 1 × 10−5 M). (b) The locally enlarged UV-vis spectra. (c) Fluorescence spectra of BNP-PX and BNP-PA in toluene (concentration: 1 × 10−5 M). (d) Cyclic voltammograms of BNP-PX and BNP-PA measured in CH2Cl2 (0.1 M n-Bu4NPF6) at the scan rate of 0.1 V/s. (e) Fluorescence spectra of BNP-PX in THF − water mixtures (8.0 × 10−4 M, excited at 424 nm) with varied volumetric fractions (f
w) of water. (f) The fluorescence intensity ratios (I/I
0) of BNP, BNP-PX, and BNP-PA in different fractions of water (I
0 is the fluorescence intensity of BNP, BNP-PX, and BNP-PA in THF). (g) The digital photo of BNP-PX in THF − water mixtures under UV light (8.0 × 10−4 M, excited at 365 nm).
Figure 3 (a) UV-vis absorption spectra of BNP-PX and BNP-PA in toluene (concentration: 1 × 10−5 M). (b) The locally enlarged UV-vis spectra. (c) Fluorescence spectra of BNP-PX and BNP-PA in toluene (concentration: 1 × 10−5 M). (d) Cyclic voltammograms of BNP-PX and BNP-PA measured in CH2Cl2 (0.1 M n-Bu4NPF6) at the scan rate of 0.1 V/s. (e) Fluorescence spectra of BNP-PX in THF − water mixtures (8.0 × 10−4 M, excited at 424 nm) with varied volumetric fractions (f
w) of water. (f) The fluorescence intensity ratios (I/I
0) of BNP, BNP-PX, and BNP-PA in different fractions of water (I
0 is the fluorescence intensity of BNP, BNP-PX, and BNP-PA in THF). (g) The digital photo of BNP-PX in THF − water mixtures under UV light (8.0 × 10−4 M, excited at 365 nm).
The electrochemical behaviors of BNP-PX and BNP-PA in anhydrous DCM were investigated by means of cyclic voltammetry (CV) as depicted
in [Figure 3d]. BNP-PX displayed a reversible reduction process at the half-wave potential of −1.88 V vs.
Fc+/Fc ([Figure 3d]). In addition, one reversible, one quasi-reversible, and one irreversible oxidation
processes were observed with a half-wave potential at 0.38, 0.87, and 1.28 V vs. Fc+/Fc ([Figure 3d]), respectively. For BNP-PA, one quasi-reversible reduction process was identified with a half-wave potential
at −2.06 V vs. Fc +/Fc ([Figure 3d]). Moreover,BNP-PA exhibited one reversible and one irreversible oxidation processes with a half-wave
potential at −0.28 and 0.52 V vs. Fc +/Fc ([Figure 3d]), respectively. However, under high potential (>0.8 V vs. Fc +/Fc), the CV curves
of BNP-PA became complex owing to its low electrochemical stability ([Figure S54]). Accordingly, the LUMO energy levels are estimated to be −2.92 and −2.74 eV for
BNP-PX and BNP-PA, respectively ([Table S3]). On the basis of their optical energy gaps obtained from their absorption edges,
the HOMOs of BNP-PX and BNP-PA are derived to be −5.42 and −5.22 eV, respectively ([Table S3]). The HOMO and LUMO energy levels of BNP-PX and BNP-PA are obviously higher than that of BNP (HOMO: −6.06 eV; LUMO: −3.50 eV), manifesting the important role of the integrated
donor moieties (phenoxazine and phenylphenazine groups). In addition, the experimental
results are well supported by the DFT calculations ([Figure 4] and [Table S3]). The HOMOs in BNP-PX and BNP-PA are mainly localized on the phenoxazine and phenylphenazine groups due to their electron-donating
properties. In contrast, the LUMOs of BNP-PX and BNP-PA are fully localized over the BNP motif. In comparison with BNP ([Figure S56]), the frontier molecular orbitals of BNP-PX and BNP-PA are spatially separated, indicating their typical D–A configurations.[5a]
[17]
 Figure 4 Calculated molecular orbitals and energy diagrams of (a) BNP-PX and (b) BNP-PA.
Figure 4 Calculated molecular orbitals and energy diagrams of (a) BNP-PX and (b) BNP-PA.
The thermal stability of BNP-PX was analyzed by the thermogravimetric analysis (TGA) method under a N2 atmosphere, as shown in [Figure S55]. At the beginning of the heating process (25–150 °C), the adsorbed water or residual
organic solvent is released, resulting in a weight loss of ∼2.0%. At the temperature
range of 150–280 °C, there is a weight loss of ∼3.0% occurred on the TGA curve, suggesting
the decomposition of BNP-PX. Accordingly, BNP-PX exhibits a decomposition temperature T
d (T
d5%, corresponding to 5% weight loss) more than 280 °C, indicating its relatively good
thermal stability. To evaluate the electroluminescence (EL) behavior of BNP-PX as the emitter in the host–guest and nondoped systems, vacuum-sublimated OLEDs with
a configuration of indium tin oxide (ITO)/MoO3 (10 nm)/1,1′-bis[4-(di-p-tolylamino)phenyl]-cyclohexane (TAPC) (30 nm)/4,4′,4″-tri(N-carbazolyl)-triphenylamine
(TCTA) (5 nm)/emissive layer/1,3,5-tri(m-pyridin-3-ylphenyl)benzene (TmPyPB) (65 nm)/LiF
(1 nm)/Al (100 nm) were fabricated, in which MoO3, TAPC, and TmPyPB worked as the hole-injection, hole-transporting, and electron-transporting
layers, respectively ([Figure 5a]). In addition, TCTA and LiF were used as the exciton-blocking layer and the electron-injection
layer, respectively ([Figure 5a]). As shown in [Figure 5 (b, c)], the turn-on voltage of the devices is around 6.0 V, and the maximum brightness
is 317 cd/m2. Varying the driving voltages of 6.0, 8.0, and 10.0 V, the devices show very small
spectra shifts ([Figure S62]). The EL spectra and color coordinates of these devices show a small variation from
low-to-high driving voltages, implying the efficient energy transfer from the host
to the guest. Among the devices fabricated, the optimized device exhibits a maximum
external quantum efficiency (EQE) of 0.6% and a maximum current efficiency of 1.0
cd/A ([Figure 5d]).
 Figure 5 (a) Schematic diagrams of the basic structures of the OLED device based on BNP-PX. (b) Current density − voltage − luminance (J − V − L) characteristics. (c) Current efficiency (CE) − luminance characteristics. (d) External
quantum efficiency (EQE) − luminance characteristics.
Figure 5 (a) Schematic diagrams of the basic structures of the OLED device based on BNP-PX. (b) Current density − voltage − luminance (J − V − L) characteristics. (c) Current efficiency (CE) − luminance characteristics. (d) External
quantum efficiency (EQE) − luminance characteristics.
Conclusions
In conclusion, we report the synthesis of two novel BN-coordinated phenalene derivatives
containing D–A structures (BNP-PX and BNP-PA). Single-crystal analysis together with the DFT calculations reveals that both BNP-PX and BNP-PA possess highly twisted geometries, which render the spatial separation of the HOMO
and LUMO distributions. By introduction of the phenoxazine and phenylphenazine substituents,
the frontier orbital energy levels of BNP-PX (HOMO: −5.42 eV; LUMO: −2.92 eV) and BNP-PA (HOMO: −5.22 eV; LUMO: −2.74 eV) are increased compared with the parent BNP (HOMO: −6.06 eV; LUMO: −3.50 eV). In addition, BNP-PX demonstrates excellent AIE effect and mega-large Stokes shift (8,033 cm−1) among all the reported BN-coordinated compounds. Furthermore, the OLED device was
fabricated based on BNP-PX with the EQE up to 0.6%. This work reported herein demonstrates that the B–N-coordinated
PAHs with donor–acceptor configurations can be generally interesting for optoelectronic
devices.




