

Subscribe to RSS
DOI: 10.1055/s-0042-1748866
Larotrectinib as an Effective Therapy in Congenital Infantile Fibrosarcoma: Report of Two Cases
Authors

Abstract
Congenital infantile fibrosarcoma (CIF) is a rare tumor in children that occurs in the first years of life. It usually arises in the extremities but some cases affect the trunk, neck, abdomen, or retroperitoneum. Surgical resection has been traditionally the treatment of choice but the development of genomic analysis and targeted therapies has shed light on new therapeutic options.
We present two patients with a congenital mass, one in the abdominal cavity (1-month-old) and the second in the left lower extremity respectively (2-months-old). In both cases, the clinical and radiological findings showed heterogeneous masses with rapidly progressive growth. MRI in the first patient exhibited an abdominal mass surrounding the aorta and inferior vena cava associated with a giant infrarenal aortic aneurysm. CT-guided biopsy was performed with pathological findings of fibrosarcoma and ETV6-NTRK3 gene fusion. The second patient underwent open biopsy also with histopathological diagnosis of fibrosarcoma and the same mutation in the TRK gene (NTRK3). Targeted therapy with a specific TRK inhibitor, larotrectinib, was started in both patients. Periodical controls were made by ultrasound or MRI, and after a few weeks of treatment, both children showed significant decrease in the mass. By the second and third months after starting the treatment, both tumors disappeared. The first patient is now 15-months-old and the second one is 8-months-old.
Larotrectinib is a novel targeted therapy with excellent results in CIF but long-term outcomes are limited to establish it as a gold standard treatment.
The mainstay treatment of congenital infantile fibrosarcoma is surgical excision but the new target therapies could make this change by avoiding morbidity and mortality associated with surgical treatment.
Introduction
Congenital infantile fibrosarcoma (CIF) is a rare tumor that arises from soft tissue. Its incidence is 5 cases per 1,000,000 infants and mainly arises from the extremities and the trunk.[1] [2] [3] The NTRK mutation seems to be the cause of tumor development, the most predominant being the ETV6-NTRK3 gene variant. It has a good prognosis having 5-year overall survival rates of 90%. Surgery remains the gold standard treatment but when it is not feasible, neo-adjuvant chemotherapy maybe effective.[1] [3] [4] Recently targeted therapies had been used to treat locally advanced and chemotherapy-resistant sarcomas with good results.[5] [6] [7] We report two cases of non-surgical treatment in two neonates.
Case Report
Case 1
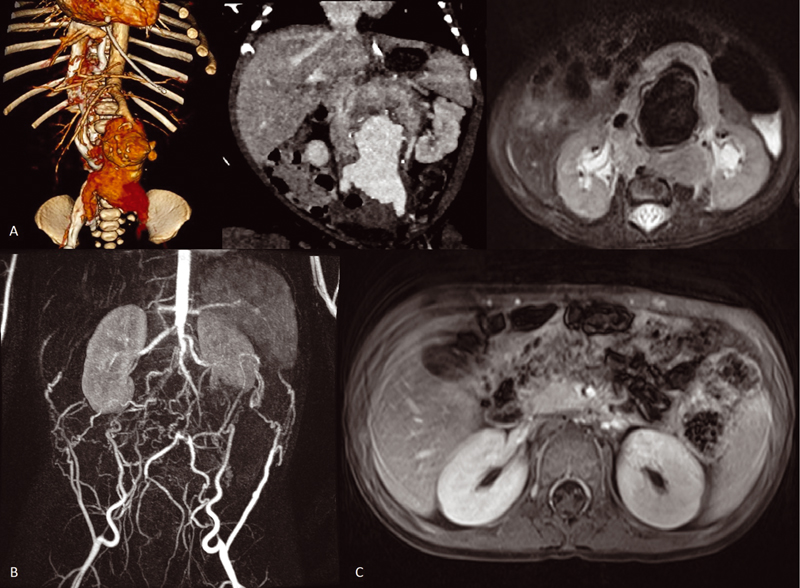
A female newborn at 40-week gestational age with prenatal diagnosis at 35 + 4 gestational age of abdominal vascular anomaly was referred for evaluation. Physical examination revealed an abdominal mass. An MRI and angio-CT were performed with suggestive findings of a retroperitoneal mass encompassing major abdominal vessels and aneurysmatic dilatation of the abdominal aorta ([Fig. 1]) with measures of 6.4 × 4.8 × 9.4 cm. After a CT-guided biopsy, pathological results were consistent with ETV6-NTRK3 mutated CIF and treatment with specific TRK inhibitor (larotrectinib) was initiated at age of 2 months at a dose of 100 mg/m2 twice daily for 1 year. Excellent response to the treatment was observed with total regression of the retroperitoneal mass by the third month after onset treatment ([Fig. 1]). Despite total regression of the mass, the abdominal aneurism grew, so surgical excision with infra renal aortic ligation was made after an occlusion test with good tolerance. The patient is now 15 months old and is in her seventh month without treatment with no radiological or clinical evidence of tumor recurrence doing normal life.
Case 2
A 2-month-old male child was referred to our institution with a lower extremity mass, which had started to grow in the first month of life. An MRI was made after an ultrasound with suggestive findings of possible hematoma. MRI revealed a soft tissue mass (5.1 × 5.6 × 6.9 cm) compatible with possible vascular malformation without being able to rule out other possibilities such as rhabdomyosarcoma ([Fig. 2]), so an open biopsy was made. During the biopsy, intensive bleeding was observed. Anatomopathological findings were conclusive with CIF and ETV6-NTRK3 translocation. Larotrectinib was initiated at 3 months of age at a dosage of 100 mg/m2 twice daily. Periodical US controls were made showing the total remission of the tumor by the second month of treatment ([Fig. 2]). The patient is actually on his eighth month of treatment without any evidence of radiological tumor recurrence.

Discussion
Despite being a rare tumor, CIF treatment has great survival rates and despite its high morbidity associated with surgical excision, which remains the gold standard treatment. When radical resection is unfeasible, neo-adjuvant chemotherapy using vincristine, actinomycin, and cyclophosphamide shows responses, allowing the posterior excision of the tumor.[1] [4] Other reported cases where resistant to the treatment or had serious complications as neutropenia.[7] [8]
Recently, genomic studies have been conducted allowing to know the molecular origin for these tumors. Translocation t(12;15)(p13;q25) with the transcript ETV6-NTRK3 is a recurrent mutation in this tumor; so, genetic studies should be done in all these patients. Targeted therapy for this mutation had great results in adult's sarcomas; however, there was no evidence of effectiveness in children until the first open-label, multicentric study was done.[6] This phase 1 study demonstrated that larotrectinib was safe and induced tumor sustained regression in over 90% of children with the TRK gene mutations and could recommend a dose of 100 mg/m2.
Since the publication of this study, a few more cases have been published. The first ones only had partial remission of the tumor allowing posterior surgical treatment[5] but posterior works published complete response to the treatment.[2] [7]
Those findings, supported by our two cases, shed light on the treatment of CIF, avoiding surgical treatment with a rapid decrease in the tumor size. More studies are needed but the effectiveness and safety of larotrectinib can make it as the targeted therapy in the mainstay treatment of TRK mutated tumors.
Conclusion
Larotrectinib is a novel targeted therapy with excellent results in CIF but long-term outcomes are limited to establish it as a gold standard treatment.
Conflict of Interest
None declared.
-
References
- 1 Ferrari A, Orbach D, Sultan I, Casanova M, Bisogno G. Neonatal soft tissue sarcomas. Semin Fetal Neonatal Med 2012; 17 (04) 231-238
- 2 Caldwell KJ, De La Cuesta E, Morin C, Pappo A, Helmig S. A newborn with a large NTRK fusion positive infantile fibrosarcoma successfully treated with larotrectinib. Pediatr Blood Cancer 2020; 67 (09) e28330
- 3 Orbach D, Rey A, Cecchetto G. et al. Infantile fibrosarcoma: management based on the European experience. J Clin Oncol 2010; 28 (02) 318-323
- 4 Corral Sánchez MD, Galán Gómez V, Sastre Urgelles A. et al. Treatment of infantile fibrosarcoma associated to an abdominal aortic aneurysm with larotrectinib: a case report. Pediatr Hematol Oncol 2021; 38 (05) 504-509
- 5 DuBois SG, Laetsch TW, Federman N. et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer 2018; 124 (21) 4241-4247
- 6 Laetsch TW, DuBois SG, Mascarenhas L. et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 2018; 19 (05) 705-714
- 7 Bielack SS, Cox MC, Nathrath M. et al. Rapid, complete and sustained tumour response to the TRK inhibitor larotrectinib in an infant with recurrent, chemotherapy-refractory infantile fibrosarcoma carrying the characteristic ETV6-NTRK3 gene fusion. Ann Oncol 2019; 30 (Suppl_8, Suppl_8) i31, i35
- 8 Edwards TM, Duran MS, Meeker TM. Congenital infantile fibrosarcoma in the premature infant. Adv Neonatal Care 2017; 17 (06) 440-450
Address for correspondence
Publication History
Received: 17 July 2021
Accepted: 18 January 2022
Article published online:
25 June 2022
© 2022. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Ferrari A, Orbach D, Sultan I, Casanova M, Bisogno G. Neonatal soft tissue sarcomas. Semin Fetal Neonatal Med 2012; 17 (04) 231-238
- 2 Caldwell KJ, De La Cuesta E, Morin C, Pappo A, Helmig S. A newborn with a large NTRK fusion positive infantile fibrosarcoma successfully treated with larotrectinib. Pediatr Blood Cancer 2020; 67 (09) e28330
- 3 Orbach D, Rey A, Cecchetto G. et al. Infantile fibrosarcoma: management based on the European experience. J Clin Oncol 2010; 28 (02) 318-323
- 4 Corral Sánchez MD, Galán Gómez V, Sastre Urgelles A. et al. Treatment of infantile fibrosarcoma associated to an abdominal aortic aneurysm with larotrectinib: a case report. Pediatr Hematol Oncol 2021; 38 (05) 504-509
- 5 DuBois SG, Laetsch TW, Federman N. et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer 2018; 124 (21) 4241-4247
- 6 Laetsch TW, DuBois SG, Mascarenhas L. et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 2018; 19 (05) 705-714
- 7 Bielack SS, Cox MC, Nathrath M. et al. Rapid, complete and sustained tumour response to the TRK inhibitor larotrectinib in an infant with recurrent, chemotherapy-refractory infantile fibrosarcoma carrying the characteristic ETV6-NTRK3 gene fusion. Ann Oncol 2019; 30 (Suppl_8, Suppl_8) i31, i35
- 8 Edwards TM, Duran MS, Meeker TM. Congenital infantile fibrosarcoma in the premature infant. Adv Neonatal Care 2017; 17 (06) 440-450