

Subscribe to RSS
DOI: 10.1055/s-0039-1690904
The Problem with Problems: Fundamental to Applied Research Using Palladium
Authors
The studies are very grateful for the support we have received from the following sources for the chemistry described in this Account: EPSRC (EP/R025754/1), The Leverhulme Trust (RPG-2015-308), and GlaxoSmithKline and AstraZeneca for several CASE awards and iCASE studentships.
Publication History
Received: 18 February 2020
Accepted after revision: 01 April 2020
Publication Date:
05 May 2020 (online)

Dedicated to our co-workers and collaborators, past, present, and future
Abstract
This Account describes the development of our cross-coupling and medicinal chemistry research from its origins at the outset of my independent career through to the present day. Throughout, the decisions and motivations as well as the mistakes and pitfalls are discussed.
1 Introduction
2 Hobbies and Interests
3 Scooped before Starting
4 Opportunity from Collaboration
5 Chemoselective Suzuki–Miyaura Cross-Coupling
6 Applications to the Medicinal Chemistry Campaign
7 Autotaxin and Heterocycle Synthesis
8 ATX Hybrids and Pd(II) Speciation
9 Conclusions
Biographical Sketches

Allan obtained his MSc degree from the University of Strathclyde, Glasgow, where he continued for his PhD studies in organometallic methodology. He then moved to a postdoctoral fellow position at Princeton University working with Professor David W. C. MacMillan on natural product total synthesis. In 2010 he returned to the UK to an industrial postdoctoral position at GlaxoSmithKline and in 2011 started his independent career at the University of Strathclyde, moving to the University of St Andrews in January 2018. His research interests are based around the development of new catalytic processes and their application in medicinal chemistry and agrochemistry.
Introduction
‘Scientists are generally good at finding solutions to problems. The biggest problem with starting a research program is identifying a good problem.’ This is the piece of advice that I remember most from the time when I was thinking of proposals for my own independent career. It is fair to say that this is also, ironically, the piece of advice that I found the most problematic – pick a good problem. I’m fairly sure this might resonate with some readers. Gauging a ‘good’ problem is an entirely subjective process and how good a problem is, or more accurately, how a scientific output is valued, varies enormously. With regards to my own field, synthetic chemistry, something that might seem ‘incremental’ to an academic might be transformative for an end user across disciplines (e.g., biologist). Equally, a major conceptual or fundamental advance might be completely irrelevant from a practical perspective. The escalating problems of trial by impact factor as well as access to and polarization of funding adds a further problem to the mix and further pressure on new principal investigators (PIs). The decision of what to base an independent research program upon is absolutely a problem.
At the time of writing, our group has been operating for around eight years, which is, in my opinion, short enough to remember the difficulties at the outset and long enough to be able to reflect a little on some of the decisions and especially the failures, which, I would argue, is more valuable. In this Account I’ll try to give an overview of some of the projects we have been involved in, with some of the decisions that affected the development of the research.
I would like to highlight at the outset that all of the work reported here, as well as the wider catalogue of research we have generated over the past eight years, is the product of a series of very talented undergraduate, graduate, and postdoctoral co-workers, as well as very generous (in time, talent, and resource) collaborators in academia and industry. I have been fortunate to have been involved in this work, but the outputs described in this essay are a testament to the tenacity and ability of these co-workers and collaborators.
2
Hobbies and Interests
One of the most difficult things to do was to decide what I was interested in. At least interested in enough to write proposals on, bearing in mind that the proposal should outline the widely sought ‘program of research’. (As an aside, I find this a crazy thing to request of new PIs. If you are proposing something novel, something that has no precedent in the literature, regardless of how robust the underpinning fundamental scientific theory is, there remains a risk that it won’t work. So, outlining 5+ years of research on this untested concept seems more like an exercise in creative writing than anything else.) I enjoy a lot of different chemistry and I especially enjoy making things, and the possible utility of the arising products (I’m a big Lego fan and also worked for a long time as a professional carpenter), hence my focus on organic chemistry. The decision over what eggs to put in the specific research program basket was a difficult one, but catalysis was an area that I thought would allow our group to make possibly useful products and develop potentially useful understanding.
3
Scooped before Starting
I thought I’d include this story as I think this is a fairly common concern and/or experience for new PIs. During grad school studies, I was involved in a project that involved conjugate addition to a vinyl 1,2-4-oxadiazole[1] (Scheme [1, a]), and in my postdoc I was involved in asymmetric organocatalysis, with one aspect being enantioselective conjugate addition via iminium catalysis. The combination of these was the essence of what was my first proposal – enantioselective conjugate addition to vinyl heterocycles, which I had thought of as being based on Hayashi-type Rh catalysis using organoborons (Scheme [1, b]).[2] [3]

Disaster struck, however, in the run up to applying for positions with my newly minted set of proposals, when the first example of this asymmetric conjugate addition (reduction in this initial case) was published by Prof. Hon Lam,[4] at the time at the University of Edinburgh, now at the University of Nottingham (Scheme [1, c]).[5] Over the last 10 years or so, Prof. Lam has done remarkable things in this area using several transition metals and novel ligand architectures, developing beautiful chemistry that is significantly beyond anything I had imagined. So, a small consolation was that the chemistry ended up in the best place with the best person.
I reworked this proposal to focus on asymmetric protonation instead of conjugate addition, and we have recently published our first example of this approach (Scheme [2]).[6]

However, while it finally worked out, this journey has not been smooth or easy: the first grant proposal for this research was rejected, and it took seven years to piece together the resource to deliver the first paper. The ‘hang on in there’ advice is easier to dispense than to accept but there is, at least from the perspective of this project, some value in it. It certainly hasn’t worked out for other projects, and in terms of ‘academic growth’, I’d argue that knowing when to kill a project, regardless of how attached you may be to it, is very valuable (examples of this below).
4
Opportunity from Collaboration
Before starting out as a PI, I completed a second postdoc, this time in industry (GlaxoSmithKline, GSK). During this time, I was fortunate to meet some great scientists. Beyond the scope of this Account, we have had a long collaboration in Sustainable Chemistry with GSK and which led to a long-term collaboration with Sigma-Aldrich (now Merck KgA).[7]
To, finally, arrive at the subject of this Account, discussions with scientists at GSK led to an idea for collaboration on a medicinal chemistry project. Our collaborators were likeminded with regards to how we thought industry and academia could interact collaboratively and with the financial support of GSK and my department, we were able to resource a collaborative medicinal chemistry project.
A major target for GSK at that time was idiopathic pulmonary fibrosis (IPF). To keep this new collaboration distinct, we constructed a project towards IPF intervention via a peripheral target (i.e., not an active project at GSK).
This was initially based on development of antagonists for LPA1, a member of a family of G-protein-coupled receptors (GPCRs) termed LPA1–6. This signaling axis begins by the hydrolysis of lysophosphatidyl choline (LPC) by the enzyme autotaxin (ATX; important later) leading to lysophosphatidic acid (LPA, Scheme [3]). LPA is an agonist for LPA1–6 and leads to a signaling cascade that can promote, amongst other effects, cell migration, proliferation, and survival. Misregulation of this pathway has been implicated in several pathologies including, amongst others, autoimmune diseases, cancer, and, the therapy area for the emerging collaboration, IPF.[8]

Medicinal chemistry is an expensive pursuit and, as a new research group with limited resource, could have been a serious problem to deliver. (Another aside: no biology resource was available in our labs or at GSK, so we had to develop additional collaborations to deal with this key aspect of the project – more on this later.)
In terms of where to start, looking into the chemical and patent literature in this area, a patent by Amira pharmaceuticals stood out. AM095 and AM966 (Figure [1]) were attractive as they are simple to build, which would allow straightforward synthesis and generation of chemical matter for scaffold-hopping structure–activity relationships (SAR).[9]

Perhaps more importantly, AM095 and AM966 could be built via Suzuki–Miyaura cross-coupling. At the same time the medicinal chemistry project was beginning, we also had an interest in boron chemistry, in particular ligand exchange at boron (speciation), as well as chemoselectivity in cross-coupling. A scaffold-hopping campaign based on the Amira compounds offered the opportunity to investigate boron speciation/chemoselective Suzuki–Miyaura cross-coupling whilst targeting scaffolds of relevance to the medicinal chemistry project. From my perspective this was as close to ideal as a research program could be: it enabled fundamental catalysis to be aligned with specific application. It also allowed the catalysis-focused members of the group to engage more effectively with the medicinal chemists and vice versa, and additional benefits of maximizing resources – suitable chemical matter from the catalysis methodology work could be assessed in assays as possible LPA1 antagonists, generating additional SAR.
5
Chemoselective Suzuki–Miyaura Cross-Coupling
Using AM095 as a workhorse, retrosynthesis gives a straightforward Suzuki–Miyaura disconnection (Scheme [4]).

Alternative placements of the electrophilic and nucleophilic functional groups were of course possible, and use of protected organoboron groups was also possible if, for example, a dinucleophile was to be used in place of the dielectrophile; however, the overall challenge that this inspired remained – was it possible to perform two Suzuki–Miyaura cross-couplings in a single operation?
From the components in Scheme [4] (a), the conventional approach would be to undertake a single cross-coupling using one organoboron with the dielectrophile (Scheme [4, b], top). The dielectrophile would require a sufficient reactivity gradient between the electrophilic sites (e.g., 1-bromo-4-chlorobenzene) such that a suitable catalyst would selectively engage the most labile C–X bond. Isolation of this intermediate would then allow a second cross-coupling with a second organoboron, using a different catalyst capable of engaging the remaining, less reactive, electrophilic site.
As noted above, the challenge we identified was to be able to do all of this in a one-pot operation without any intervention, which would require simultaneous control of two electrophiles and two nucleophiles (Scheme [4, b], bottom). Electrophile chemoselectivity was well established and we assumed this would be relatively straightforward based on established reactivity patterns and knowledge of ligand effects in Pd catalysis.
The key phrase here is ‘assumed’. While it was well-known that chemoselective cross-coupling is possible with two (and more) electrophilic sites in a system, these processes only engage (i.e., react) one site at a time. Use of a single catalyst to sequentially and selectively engage two inequivalent nucleophilic sites was, to our knowledge, unprecedented. For example, in a system containing two equivalent electrophilic sites, it was possible to either promote a single cross-coupling or exhaustive cross-coupling (e.g., Scheme [5, a]).[10] [11] [12] Use of inequivalent dielectrophiles had at that time only been reported to undergo selective single cross-couplings, for example, the seminal work of Fu (Scheme [5, b]).[13] An excellent review of this area was published by Spivey and co-workers.[14]

While challenges were at least clear from the electrophile side, things were less simple from the nucleophile perspective. Cross-coupling in a system containing two organoborons, at the time, had been achieved only through the use of protecting group chemistries, and these processes again only coupled one site (the protected organoboron remaining intact). Specifically, for aryl organoborons, the use of the base-labile BMIDA has been extensively developed by Burke[15] (e.g., Scheme 6, a[16]) based on the work of Mancilla and co-workers in the 1980s,[17] or the acid-labile BDAN protecting group developed by Suginome (e.g., Scheme 6, b[18]).[19] Other excellent work in the area of selective Suzuki–Miyaura cross-coupling exploiting reactivity of alkyl diboron systems has been reported by Shibata,[20] Morken,[21] Hall,[22] Crudden,[23] and others – an excellent review of this area was published by Crudden.[24] [25]

To achieve our planned sequential cross-coupling, the system would require two organoborons that were reactive towards transmetalation. This suggested nucleophile selectivity based on kinetic discrimination at transmetalation. To this end, Hartwig demonstrated that arylboronic acids transmetalate to (HO)(aryl)Pd(II) complexes ca. 30 times faster than the equivalent arylboronic acid pinacol ester (BPin).[26] Based on this, a system containing an arylboronic acid and aryl BPin may then seem like an ideal starting point for investigating this process; however, we were aware of issues relating to speciation that seemed likely to obviate any possible kinetic advantage of the arylboronic acid. Specifically, a 1:1 mixture of boronic acid 1a and BPin 2b will rapidly equilibrate to generate two different boronic acids (1a and 2a) and their associated BPins (1b and 2b; Scheme [7]).[27] Clearly we’d have little chance of achieving selectivity if these processes took place during the desired reaction.

As a first step to address the ‘speciation problem’, we planned a reaction that allowed us to investigate whether some speciation control was possible, establishing a formal C(sp2)BPin homologation process by leveraging aspects of BMIDA chemistry (Scheme [8]). Here, an aryl BPin would be cross-coupled with a haloaryl BMIDA to generate a new aryl BPin. The process would take place by exploiting several base-promoted events, taking advantage of the usually basic reaction conditions associated with Suzuki–Miyaura chemistry. The initial cross-coupling of 3 and 4 would generate BMIDA adduct 5 as well as the byproduct from this cross-coupling, HOBPin (assuming that ArBPin undergo direct transmetalation). Base-mediated hydrolysis of both of these species would liberate the parent boronic acid 6a and pinacol. These would then undergo esterification leading to the desired product 6b. Several pieces of information suggested that this all would be feasible in principle, including Burke’s slow release of boronic acids from BMIDA species[28] and that esterification of arylboronic acids with diols at basic pH is well-known in saccharide sensing, with significant information on binding affinities, etc., available from numerous studies in this field.[29]

Despite this information, there were problems associated with this chemistry. Specifically, controlling the rate of BMIDA hydrolysis:[30] if either or both of the BMIDA starting material 4 or intermediate biaryl BMIDA 5 were to undergo hydrolysis before the Suzuki–Miyaura process was complete, oligomerization would occur. Indeed, this was the major issue faced with this methodology development;[27] [31] however, appropriate balance of the base:H2O ratio in the system allowed effective control, enabling realization of a general method for this BPin synthesis (Scheme [9], top). As a side note, the BPin process required heating to 90 °C; the same reaction conditions at room temperature allowed retention of the BMIDA, demonstrating that the BMIDA unit can tolerate basic conditions, at least in some cases (Scheme [9], bottom).[27] [32]

At the time, we knew that we needed very dry Suzuki–Miyaura conditions – 3 equiv K3PO4 and 5 equiv H2O.[27] [31a] While we documented the effects of base and H2O on this specific system, we had only empirical evidence for these effects: too little H2O led to poor cross-coupling, too much led to rapid BMIDA hydrolysis and oligomerization issues. Besides the oligomerization issue, we found that these conditions facilitated a very fast Suzuki–Miyaura reaction, which struck us as odd especially considering the that the majority of Suzuki–Miyaura reactions are often conducted using water as co-solvent or aqueous base; however, tracking down why this was the case came later (vide infra).
This initial system was a first step towards sequential cross-coupling. We had shown it was possible to use two organoborons (one protected as a BMIDA) and one electrophilic site to generate a new C–C bond as well as a new and, importantly, reactive organoboron site. With this established, we sought to combine the nucleophile control with electrophile control. The design was essentially an extension of the initial protocol (Scheme [10]).[31a]

Addition of a second, less reactive, electrophile to the system would allow (i) an initial electrophile-selective Suzuki–Miyaura event between 3 and 4 to generate a biaryl BMIDA 5, (ii) the sequence of controlled hydrolysis/esterification events to generate a new BPin 6b, and (iii) a second Suzuki–Miyaura event from the remaining electrophile and the newly formed BPin to deliver the desired selectively coupled product 8. Clearly, to achieve the electrophile control, C–X1 must be more labile than C–X2.
Based on what we had learned from the homologation process, control of the hydrolytic events was relatively straightforward from a practical sense – we had to control the base:H2O stoichiometry (interestingly, note the higher loadings of both vs. the homologation process – this was necessary otherwise the second Suzuki–Miyaura stalled). The electrophile chemoselectivity was realized purely via focused ligand screening. ‘Focused’ here meaning assessing ligands that were known to engage aryl chlorides. Ultimately, this process came to fruition, allowing a first example of sequential chemoselective Suzuki–Miyaura cross-coupling (Scheme [11]).[33]

This sequential Suzuki–Miyaura process was effective and certainly allowed construction of the two C–C bonds important to the LPA1 antagonist campaign. However, with regards to sequential cross-coupling based on control of oxidative addition and transmetalation, it was, essentially, a bit of a cheat as we continued to rely upon a protecting group strategy. This left work to be done.
Based on everything we had learned regarding controlling boron speciation by this stage, we endeavored to provide an answer. Returning to Hartwig’s observations of a faster transmetalation of ArB(OH)2 to (HO)(aryl)Pd(II) complexes vs. the equivalent ArBPin,[26] we queried whether this would be observed in a practical sense, i.e., in competition.
Charting the reaction profile (conversion into product) of a simple Suzuki–Miyaura reaction using either boronic acid 2a or the equivalent BPin 2b with bromobenzene independently (under our preferred high base, low-water Suzuki–Miyaura conditions), demonstrated that the initial rates and overall reaction profiles were more or less identical, i.e., krel ca.1 (Scheme [12, a]).[34]

However, the equivalent competition experiment under the same reaction conditions told a very different story (Scheme [12, b]): while the reactivity of boronic acids and the equivalent BPin appeared equivalent in isolation, they were notably inequivalent in competition, with the boronic acid 10a outcompeting the BPin 2b in the cross-coupling with a selectivity of ca. 8:1 with krel ca. 10 in favor of the boronic acid. This suggested that it was possible to discriminate between the two different boron species at transmetalation (again, this was based on the reasonable assumption that ArBPin undergo direct transmetalation).
A brief optimization of the reaction conditions resulted in a general process where ArB(OH)2 underwent chemoselective cross-coupling in the presence of ArBPin (Scheme [13, a]).[34]

This then allowed the natural extension of the process to deliver a chemoselective, sequential Suzuki–Miyaura cross-coupling using two reactive electrophiles and two reactive nucleophiles to give either a product pair, such as 13 and 14 (Scheme [13, b]), or a single product (Scheme [13, c]). The boronic acid selectivity was, as far as we could tell, perfect in the competition experiments (Scheme [13, a]); however, it was less than perfect in the sequential cross-coupling (Scheme [13, b] and c), where some erosion was detected. To sum up this section, these investigations showed that sequential Suzuki–Miyaura cross-coupling was possible; however, from a practical perspective, the better method is the process outlined in Scheme [11], where controlling the BMIDA hydrolysis events provide an additional control to enforce chemoselectivity.[33]
6
Applications to the Medicinal Chemistry Campaign
Beyond IPF, our group has had interests in several other areas of medicinal chemistry, including epigenetics.[32] [33] [35] As a small demonstration of utility in this area, the BMIDA-based sequential cross-coupling process was used to prepare a pan-BET bromodomain inhibitor (Scheme [14]).[33]
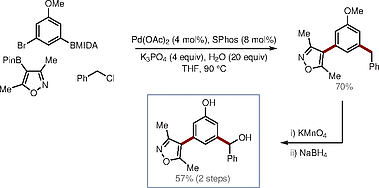
However, and rather unfortunately, despite being an inspiration for the development of the methodology, the utility of these cross-coupling methods within our LPA1 antagonist program was decidedly short lived. Guidance from our biological collaborators suggested the development of LPA1 antagonists was likely to be difficult based on high cell-surface concentrations of LPA – arising due to the chaperone effect of ATX.[8] The guidance at this stage was that ‘moving up a level’ in the signaling axis would be strategically sensible and have a greater likelihood of success. In other words, instead of competing with LPA for the GPCRs, we would be best interfering with the supply of LPA at source by developing inhibitors for ATX.
Despite this, we were successful in developing a novel LPA1 antagonist scaffold, 15, which displayed moderate potency (Figure [2]).[36] However, this was as far as the LPA1 story progressed. Throughout the process we learned a great deal from a scientific perspective – in both areas – and, from a personal viewpoint, learning to let go of a project was certainly valuable.

7
Autotaxin and Heterocycle Synthesis
Moving to target ATX was a good decision. It was a discrete molecular target and structurally enabled (i.e., there was a crystal structure and knowledge of the active site).[37] There were a series of possible assays available and, more importantly at the outset, there was patented chemical matter that, similar to the LPA1 project, gave us a reasonable place to start. In particular, there were assets from Amira[38] (16) and Pfizer[39] (PF8380) that provided interesting, and seemingly feasible, objectives (Figure [3]).

Perhaps unsurprisingly, the majority of ATX inhibitors developed at the stage we entered this area conformed to the lipid-like chemotype, i.e., they resembled the structure of the endogenous ligands LPC and LPA.[8] The Pfizer lead, PF8380, was a particularly potent compound (ca. 4 nm) and from a structural perspective, much like the criteria for selection of the starting points for the LPA1 project, this was simple to assemble. From the medicinal chemistry perspective, there was little in the way of meaningful SAR with PF8380 and no crystal structure of PF8380 bound in ATX. This latter point was significant as it formed the basis of an incredibly valuable collaboration with superb team of structural biologists at the National Cancer Institute in The Netherlands (NKI). Without the NKI team, this ATX project would not have been anywhere near as successful. Apart from providing all of the structural biology, the NKI team also delivered all of the ATX assay data. Solving the ATX-PF8380 structure was an initiation point for conversations, which led to a significant collaboration on ATX. Accordingly, PF8380 was an ideal compound to investigate in many respects.
The Amira compound 16 was a slightly different story. Significantly, the structure does not conform to the lipid-like chemotype and so it was intriguing from that perspective. Despite being reported to be a potent compound (K i <0.3 μM), the patent data was ‘binned’ into three categories – K i > 1 μM, 0.3 > K i < 1 μM, and K i < 0.3 μM – meaning there was very little by way of SAR for this compound.[38] In addition, while PF8380 was a potent compound, it suffered some significant solubility issues. Compound 16, however, offered greater solubility combined with unique structural qualities, such that we considered it significantly ‘more developable’. In other words, had a greater chance for exploration of topological space to develop our own asset based on a similar chemotype. This required SAR knowledge.
We duly undertook the unexpectedly challenging synthesis of a small library of compounds based on 16, which were evaluated in ATX assays to establish SAR.[40] The synthetic route to an exemplar compound in the library (18) is shown in Scheme [15]. The main issues with the route were (i) the Fisher indole synthesis, which was not only low yielding (around 35% overall yield) but delivered a ca. 3:1 mixture of regioisomers 17a and 17b that were difficult to resolve (and only 17b required), and (ii) the penultimate Ullmann–Goldberg step which was also very low yielding (ca. 30%).

Despite the hurdles in the synthetic campaign, a meaningful library was prepared. The SAR told us a reasonable story and, coupled with some modelling data that aligned with the assay data, this was certainly believable. However, structural biology put all of this to rest. Co-crystals of 16-ATX revealed that the compound didn’t bind in the active site but rather bound in a region called the ‘tunnel’, which is remote from the active site. Perhaps unsurprisingly, follow-up Michaelis–Menten and Lineweaver–Burk studies established that 16 was a noncompetitive inhibitor of ATX. Binding of similar compounds to the tunnel region was reported at approximately the same time by two other research groups.[41] [42]
This inarguable biological data meant that we had, once again, to walk away from a project, in this case to move on from the idea that 16 was a reasonable starting point for scaffold hopping and subsequent development.
Before moving on, I thought I’d illustrate how we planned to use 16 as a starting point for our own discovery, as this led to some new synthetic chemistry. As noted briefly above, the synthesis of the library used for SAR on 16 was not trivial. Besides the synthetic campaign around establishing SAR, we developed chemistry that would allow us to make a series of substituted indoles more easily. Functionalizing the 2-position of the indole as well as being able to control the functionality and regiochemistry around the benzenoid ring was important to our objectives. The direct C2-functionalization of indoles, in the various ways that this is possible, was not ideal for our purposes. Ultimately, we designed some chemistry based on the Cacchi reaction[43] [44] using a borylated alkyne (Scheme [16]).[7i,45]


This would allow the indole to be built, controlling the functionality on the benzenoid ring as well as the substitution (or, here, the lack of) at C3 and, in terms of added value from a general synthetic methods perspective (in our opinion), installed a BMIDA unit in the C2 position.
From the medicinal chemistry perspective, this would allow us access to a range of useful compounds fairly rapidly. However, the structural biology and kinetic data arrived while we were establishing these methods, meaning they were no longer necessary for this specific purpose. However, this didn’t diminish the general synthetic value of the method, which is an effective approach to 2-borylated indoles and benzofurans, as well as other fused heterocycles (Scheme [17, a]).[7i] [45] Apart from cross-coupling processes using the BMIDA, these compounds can be oxidized fairly easily to give access to 2-oxindoles, another important pharmacophore (Scheme [17, b]).[46]
Importantly, in addition to oxidation, the BMIDA performed the functions that would be expected. Specifically, Suzuki–Miyaura cross-coupling was straightforward to either use the BMIDA as the nucleophilic component (Scheme [18, a]) or retain in couplings of other sites (Scheme [18, b]), or during other chemistries, such as hydrogenation and Chan–Lam amination (Scheme [18, c]).[45]

8
ATX Hybrids and Pd(II) Speciation
Returning to the medicinal chemistry, the Amira project was discontinued; however, the concurrent PF8380 project started delivering some interesting results. Following preparation of PF8380 (synthetic route shown in Scheme [19]), our structural biologist collaborators solved the structure of the PF8380-ATX co-crystal, which gave an unexpected observation (Figure [4]).[47]


A percentage of the crystals were found to have a bile acid – UDCA or TUDCA – bound in the tunnel region along with PF8380 in the active site. Perhaps unsurprisingly, these bile acids are noncompetitive modulators of ATX (similar to the Amira compound 16 [40]). This offered an interesting opportunity: we believed that a novel series of ATX inhibitors could be generated through creation of a hybrid series of compounds containing elements of the bile acid and PF8380 (Scheme [20]).

We considered this approach to be attractive from three perspectives: (1) parallel studies indicated that the benzoxazolidinone head group of PF8380 was the main contributor to the physicochemical property problems of this molecule – the hybrid compounds would lack this motif; (2) since the bile acid occupies the tunnel region and the dichloroarene occupies the lipophilic pocket, it would not be possible for the enzyme to accommodate LPC (or any other sizeable ligand) possibly turning a noncompetitive modulator (the bile acid) into a competitive inhibitor; and (3) if correct, this would represent a new class of ATX inhibitor, giving some possible IP space.
To explore this, we created a series of hybrid compounds based on the bile acid joined to the dichloroarene via a series of linkers. The general synthetic route is shown in Scheme [21] alongside selected compounds from this series.[47]

This approach proved very successful: the hybrid strategy did indeed change the mode of action of the bile acids from noncompetitive to competitive and, moreover, these were potent compounds. This initial investigation formed the basis for further development studies based on this chemotype, which won’t be discussed here. However, in a last foray into the chemistry inspired by this project, we recognized a challenge in development of this series. We had some information on chemotypes that bind in the tunnel, such as Amira compound 16, related compounds disclosed at around the same time, and the bile acids; however, a systematic exploration of this region was lacking.
Sticking with our hybrid series, establishing the minimum determinants of potency was considered important to further development work. This then became an exercise in a classic problem for synthetic chemistry – how to build steroid scaffolds. Moreover, a bigger synthetic challenge was in play here – how to systematically explore SAR on steroid scaffolds.
Before launching a potentially very resource intensive synthetic chemistry project, we realized we might be able to offer some advantage by again leveraging our interests in speciation. Specifically, based on our work with the Suzuki–Miyaura reaction, we became interested in the possibility of controlling chemoselectivity by controlling Pd(II) speciation. The fundamental project we envisaged was based on selective cross-coupling of vinyl BPin (Scheme [22]).[48] [49]

Vinyl BPin is a competent nucleophile for Suzuki–Miyaura cross-coupling and can undergo Mizoroki–Heck reactions at the terminal carbon.[50] Importantly, these two processes are driven by two different Pd(II) species: the Suzuki–Miyaura reaction is driven by (Ar)Pd(II)OH complexes (oxopalladium pathway)[26] , [51] [52] [53] while the Mizoroki–Heck is driven by (Ar)Pd(II)X (where X = (pseudo)halide) and these complexes are in an equilibrium. On the fundamental level, we were interested in whether we could manipulate this equilibrium effectively to allow selectivity for Suzuki–Miyaura or Mizoroki–Heck.
Studies by Amatore and Jutand,[51] Denmark,[52] and Hartwig[26] had provided a significant body of data regarding transmetalation during Suzuki–Miyaura cross-coupling via oxopalladium species.[53] Importantly, Hartwig provided useful detail on Pd(II) anion metathesis including equilibrium constants for OH→X exchange of (R3P)2Pd(Ar)(OH) complexes.[26] Based on our previous work in boron speciation and with knowledge of the importance of H2O and base within these processes, we probed the impact of H2O and base on OH→X exchange for an exemplar Pd(II) complex (19a). A snapshot of this data is provided in Scheme [23] showing that there is also a clear impact of base and water on the equilibrium between (Ar)Pd(II)X complex 19a and (Ar)Pd(II)OH complex 19b.[48]

The data suggested, perhaps counterintuitively, that ArPd(II)OH complexes are in higher concentration under drier conditions (i.e., with less H2O in the reaction mixture), which was in agreement with Hartwig’s data.[26] From our perspective this was significant and assisted in making sense of some previous observations in our boron speciation work. Lloyd-Jones had previously established that the boronic acid–boronate equilibrium favors the boronic acid when the system is drier.[54] Accordingly, a drier reaction favors both the neutral boronic acid and the Pd(II)OH complex, both of which are required for transmetalation via the oxopalladium pathway. This was the origin of the empirical observations from our boron speciation work, where dry conditions seemed to favor a ‘fast’ Suzuki–Miyaura reaction.
Optimization allowed us to develop a system where chemoselective cross-coupling of vinyl BPin was achieved using the same reaction conditions by variation of the base – K3PO4 for Suzuki–Miyaura and Et3N for Mizoroki–Heck (Scheme [24]).[48]

From the synthetic perspective, we realized that if we could achieve this in a reaction containing three different olefins then we might be able to deliver a system that allows selective cross-coupling of vinyl BPin to deliver a diene that would undergo Diels–Alder reaction to give cyclohexenes (Scheme [25]).[48] [55] Depending on whether the reaction proceeded via Mizoroki–Heck or Suzuki–Miyaura, the cyclohexene product would be borylated or nonborylated, respectively.

In the context of the steroid SAR, this wouldn’t answer that question; however, this approach offered the possibility of generating a lot of information by allowing access to a library of carbocycles, with broad variation of structure, that we could use to explore the binding more broadly – essentially a hit finding approach.
The physical-organic aspects of the Pd project are not discussed here, but the process was successful, and we were able to develop the desired chemoselective cross-coupling/Diels–Alder processes to deliver a small library of exemplar compounds (Scheme [26]).[48]

As shown in Scheme [26], the structural diversity offered by this approach was fairly broad and importantly, the borylated compounds could be functionalized in the expected ways (e.g., Scheme [27]). We hope to report on the outcome of these studies, in terms of the impact this chemistry has had on our ATX program, in due course.

9
Conclusions
Hopefully, I have managed to take you on an honest tour of how this research came to be and the twists and turns of a new PI. Perhaps the biggest retrospective question, based on the opening sentences, is ‘Did we pick a good problem?’ I have no idea. We picked a problem: a problem we were interested in, a problem that we thought allowed us to explore as much of what we were interested in and within the limits applied by the resources we had, and tried as best we could to navigate our way through the issues we faced on the way. I do believe we have offered something useful in two main areas – new knowledge in boron chemistry and Pd catalysis as well as in the medicinal chemistry area. We – and personally speaking, I – learned a lot. A lot about various aspects of science, collaboration, project management, and more. Would – or should – we have pursued anything differently? Maybe. It’s probably natural to suggest that, if given another go, we would have avoided making mistakes. The question then becomes would we have learned as much if we didn’t make these mistakes? I very much doubt it.
Acknowledgment
For the research described here, I am extremely grateful to the following co-workers and collaborators (listed alphabetically). The research group co-workers: David Cain, Dr Nicola Caldwell, Dr Peter Campbell, Dr Diana Castagna, Dr Emma Duffy, Dr Paloma Engel-García, Dr Jamie Fyfe, Dr Rob Law, Dr Donna MacMillan, Dr Fiona McGonagle, Calum McLaughlin, Dr Lisa Miller, Dr John Molloy, Jenna Mowat, Dr Calum Muir, Dr Francis Potjewyd, Dr Ciaran Seath, Dr Elena Valverde, Dr Julien Vantourout, Matthew West, Dr Kirsty Wilson, Dr Chao Xu.
Our collaborators: Dr Niall Anderson (GSK), Prof. Glenn Burley (University of Strathclyde), Dr Neil Fazakerley (GSK), Dr David Hirst (GSK), Dr Albert Isidor-Llobet (GSK), Dr Craig Jamieson (University of Strathclyde), Dr Alan Kennedy (University of Strathclyde), Prof. Willem-Jan Keune (The Netherlands Cancer Institute), Dr Andrew Leach (University of Manchester), Dr Simon Macdonald (GSK), Prof. Andrew Morris and research group (Lexington Veterans Affairs Medical Center), Dr David Nelson (University of Strathclyde), Prof. Anastassis Perrakis and research team (The Netherlands Cancer Institute), John Pritchard (GSK), Dr Jane Murray (Merck KgA), Dr Joanna Redmond (GSK), Dima Semaan (University of Strathclyde), Dr Ian Simpson (AstraZeneca), Dr Katherine Wheelhouse (GSK), Dr Louise Young (University of Strathclyde).
-
References
- 1 Burns AR, Kerr JH, Kerr WJ, Passmore J, Paterson LC, Watson AJ. B. Org. Biomol. Chem. 2010; 8: 2777
- 2a Hayashi T, Yamasaki K. Chem. Rev. 2003; 103: 2829
- 2b Jean M, Casanova B, Gnoatto S, van de Weghe P. Org. Biomol. Chem. 2015; 13: 9168
- 3 For some of our collaborative medicinal chemistry work in Rh-catalyzed 1,4-addition, see: Anderson NA, Fallon BJ, Valverde E, MacDonald SJ. F, Pritchard JM, Suckling CJ, Watson AJ. B. Synlett 2012; 23: 2817
- 4 Rupnicki L, Saxena A, Lam HW. J. Am. Chem. Soc. 2009; 131: 10386
- 5 For a review, see: Best D, Lam HW. J. Org. Chem. 2014; 79: 831
- 6 Xu C, Muir CW, Leach AG, Kennedy AR, Watson AJ. B. Angew. Chem. Int. Ed. 2018; 57: 11374
- 7a MacMillan DS, Murray J, Sneddon HF, Jamieson C, Watson AJ. B. Green Chem. 2012; 14: 3016
- 7b MacMillan DS, Murray J, Sneddon HF, Jamieson C, Watson AJ. B. Green Chem. 2013; 15: 596
- 7c Caldwell N, Jamieson C, Simpson I, Watson AJ. B. ACS Sustain. Chem. Eng. 2013; 1: 1339
- 7d McGonagle FI, MacMillan DS, Murray J, Sneddon HF, Jamieson C, Watson AJ. B. Green Chem. 2013; 15: 1159
- 7e McGonagle FI, Sneddon HF, Jamieson C, Watson AJ. B. ACS Sustain. Chem. Eng. 2014; 2: 523
- 7f Caldwell N, Campbell PS, Jamieson C, Potjewyd F, Simpson I, Watson AJ. B. J. Org. Chem. 2014; 79: 9347
- 7g Caldwell N, Jamieson C, Simpson I, Watson AJ. B. Chem. Commun. 2015; 51: 9495
- 7h Eastman HE, Jamieson C, Watson AJ. B. Aldrichimica Acta 2015; 48: 51
- 7i Wilson KL, Kennedy AR, Murray J, Greatrex B, Jamieson C, Watson AJ. B. Beilstein J. Org. Chem. 2016; 12: 2005
- 7j McPherson CG, Caldwell N, Jamieson C, Simpson I, Watson AJ. B. Org. Biomol. Chem. 2017; 15: 3507
- 7k Wilson KL, Murray J, Sneddon HF, Wheelhouse KM. P, Watson AJ. B. Chem 2017; 3: 365
- 7l Wilson KL, Murray J, Jamieson C, Watson AJ. B. Synlett 2018; 29: 650
- 7m McPherson CG, Cooper AK, Bubliauskas A, Mulrainey P, Jamieson C, Watson AJ. B. Org. Lett. 2017; 19: 6736
- 7n Wilson L, Murray J, Jamieson C, Watson AJ. B. Org. Biomol. Chem. 2018; 16: 2851
- 7o Wilson KL, Murray J, Sneddon HF, Jamieson C, Watson AJ. B. Synlett 2018; 29: 2293
- 8a Barbayianni E, Magrioti V, Moutevelis-Minakakis GP, Kokotos G. Expert Opin. Ther. Pat. 2013; 23: 1123
- 8b Castagna D, Budd DC, Macdonald SJ. F, Jamieson C, Watson AJ. B. J. Med. Chem. 2016; 59: 5604
- 9a Swaney JS, Chapman C, Correa LD, Stebbins KJ, Bundey RA, Prodanovich PC, Fagan P, Baccei CS, Santini AM, Hutchinson JH, Seiders TJ, Parr TA, Prasit P, Evans JF, Lorrain DS. Br. J. Pharmacol. 2010; 160: 1699
- 9b Swaney JS, Chapman C, Correa LD, Stebbins KJ, Broadhead AR, Bain G, Santini AM, Darlington J, King CD, Baccei CS, Lee C, Parr TA, Roppe JR, Seiders TJ, Ziff J, Prasit P, Hutchinson JH, Evans JF, Lorrain DS. J. Pharmacol. Exp. Ther. 2011; 336: 693
- 10 Sinclair DJ, Sherburn MS. J. Org. Chem. 2005; 70: 3730
- 11 Dong C.-G, Hu Q.-S. J. Am. Chem. Soc. 2005; 127: 10006
- 12 Groombridge BJ, Goldup SM, Larrosa I. Chem. Commun. 2015; 51: 3832
- 13 Littke AF, Dai C, Fu GC. J. Am. Chem. Soc. 2000; 122: 4020
- 14 Almond-Thynne J, Blakemore DC, Prydeb DC, Spivey AC. Chem. Sci. 2017; 8: 40
- 15a Gillis EP, Burke MD. Aldrichimica Acta 2009; 42: 17
- 15b Li J, Grillo AS, Burke MD. Acc. Chem. Res. 2015; 48: 2297
- 16 Lee SJ, Gray KC, Paek JS, Burke MD. J. Am. Chem. Soc. 2008; 130: 466
- 17 Mancilla T, Contreras R, Wrackmeyer B. J. Organomet. Chem. 1986; 307: 1
- 18 Noguchi H, Hojo K, Suginome M. J. Am. Chem. Soc. 2007; 129: 758
- 19a Noguchi H, Shioda T, Chou C.-M, Suginome M. Org. Lett. 2008; 10: 377
- 19b Iwadate N, Suginome M. Org. Lett. 2009; 11: 1899
- 19c Iwadate N, Suginome M. Chem. Lett. 2010; 39: 558
- 19d Iwadate N, Suginome M. J. Am. Chem. Soc. 2010; 132: 2548
- 19e Ihara H, Koyanagi M, Suginome M. Org. Lett. 2011; 13: 2662
- 19f Feng X, Jeon H, Yun J. Angew. Chem. Int. Ed. 2013; 52: 3989
- 19g Yoshida H, Takemoto Y, Takaki K. Chem. Commun. 2015; 51: 6297
- 20 Endo K, Ohkubo T, Hirokami M, Shibata T. J. Am. Chem. Soc. 2010; 132: 11033
- 21a Mlynarski SN, Schuster CH, Morken JP. Nature 2014; 505: 386
- 21b Sun C, Potter B, Morken JP. J. Am. Chem. Soc. 2014; 136: 6534
- 21c Potter B, Szymaniak AA, Edelstein EK, Morken JP. J. Am. Chem. Soc. 2014; 136: 17918
- 22a Lee JC. H, McDonald R, Hall DG. Nature Chem. 2011; 3: 894
- 22b Sun H.-Y, Kubota K, Hall DG. Chem. Eur. J. 2015; 21: 19186
- 23a Crudden CM, Ziebenhaus C, Rygus JP. G, Ghozati K, Unsworth PJ, Nambo M, Both S, Hutchinson M, Laberge VS, Maekawa Y, Imao D. Nat. Commun. 2016; 7: 11065
- 23b Glasspoole BW, Oderinde MS, Moore BD, Antoft-Finch A, Crudden CM. Synthesis 2013; 45: 1759
- 24 Rygus JP. G, Crudden CM. J. Am. Chem. Soc. 2017; 139: 18124
- 26 Carrow BP, Hartwig JF. J. Am. Chem. Soc. 2011; 133: 2116
- 27 Fyfe JW. B, Valverde E, Seath CP, Kennedy AR, Redmond JM, Anderson NA, Watson AJ. B. Chem. Eur. J. 2015; 24: 8951
- 28 Knapp DM, Gillis EP, Burke MD. J. Am. Chem. Soc. 2009; 131: 6961
- 29a Babcock L, Pizer R. Inorg. Chem. 1980; 19: 56
- 29b Pizer R, Tihal CA. Polyhedron 1996; 15: 3411
- 29c Springsteen G, Wang B. Tetrahedron 2002; 58: 5291
- 29d Bosch LI, Fyles TM, James TD. Tetrahedron 2004; 60: 11175
- 29e Yan J, Springsteen G, Deeter S, Wang B. Tetrahedron 2004; 60: 11205
- 29f Iwatsuki S, Nakajima S, Inamo M, Takagi HD, Ishihara K. Inorg. Chem. 2007; 46: 354
- 29g Miyamoto C, Suzuki K, Iwatsuki S, Inamo M, Takagi HD, Ishihara K. Inorg. Chem. 2008; 47: 1417
- 29h Martínez-Aguirre MA, Villamil-Ramos R, Guerrero-Alvarez JA, Yatsmirsky AK. J. Org. Chem. 2013; 78: 4674
- 29i Watanabe E, Miyamoto C, Tanaka A, Iizuka K, Iwatsuki S, Inamo M, Takagi HD, Ishihara K. Dalton Trans. 2013; 42: 8446
- 29j Okamoto T, Tanaka A, Watanabe E, Miyazaki T, Sugaya T, Iwatsuki S, Inamo M, Takagi HD, Odani A, Ishihara K. Eur. J. Inorg. Chem. 2014; 2389
- 29k Furikado Y, Nagahata T, Okamoto T, Sugaya T, Iwatsuki S, Inamo M, Takagi HD, Odani A, Ishihara K. Chem. Eur. J. 2014; 20: 13194
- 30 Gonzalez JA, Maduka Ogba O, Morehouse GF, Rosson N, Houk KN, Leach AG, Cheong PH. Y, Burke MD, Lloyd-Jones GC. Nat. Chem. 2016; 8: 1067
- 31a Fyfe JW. B, Seath CP, Watson AJ. B. Angew. Chem. Int. Ed. 2014; 53: 12077
- 31b Molloy JJ, Law RP, Fyfe JW. B, Seath CP, Hirst DJ, Watson AJ. B. Org. Biomol. Chem. 2015; 13: 3093
- 32 The formal homologation also operates well for arylboronic acids, see: Muir CW, Vantourout JC, Isidro-Llobet A, Macdonald SJ. F, Watson AJ. B. Org. Lett. 2015; 17: 6030
- 33 Seath CP, Fyfe JW. B, Molloy JJ, Watson AJ. B. Angew. Chem. Int. Ed. 2015; 54: 9976
- 34 Fyfe JW. B, Fazakerley NJ, Watson AJ. B. Angew. Chem. Int. Ed. 2017; 56: 1249
- 35 Law RP, Atkinson SJ, Bamborough P, Chung C, Demont EH, Gordon LJ, Lindon M, Prinjha RK, Watson AJ. B, Hirst DJ. J. Med. Chem. 2018; 61: 4317
- 36 Castagna D, Duffy EL, Semann D, Young LC, Pritchard JM, MacDonald SJ. F, Budd DC, Jamieson C, Watson AJ. B. Med. Chem. Commun. 2015; 6: 1149
- 37a Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, Albers HM. H. G, van Meeteren LA, Houben AJ. S, van Zeijl L, Jansen S, Andries M, Hall T, Pegg LE, Benson TE, Kasiem M, Harlos K, Kooi CW. V, Smyth SS, Ovaa H, Bollen M, Morris AJ, Moolenaar WH, Perrakis A. Nat. Struct. Mol. Biol. 2011; 18: 198
- 37b Nishimasu H, Okudaira S, Hama K, Mihara E, Dohmae N, Inoue A, Ishitani R, Takagi J, Aoki J, Nureki O. Nat. Struct. Mol. Biol. 2011; 18: 205
- 37c Stein AJ, Bain G, Prodanovich P, Santini AM, Darlington J, Stelzer NM. P, Sidhu RS, Schaub J, Goulet L, Lonergan D, Calderon I, Evans JF, Hutchinson JH. Mol. Pharmacol. 2015; 88: 982
- 38a Roppe JR, Parr TA, Hutchinson JH. US 20130029948 A1, 2013
- 38b Hutchinson JH, Parr TA, Roppe JF, Stock NS, Volkots D. WO 2012024620 A2, 2012
- 38c Roppe JR, Parr TA, Hutchinson JH. WO 2012166415, 2012 .
- 39 Gierse J, Thorarensen A, Beltey K, Bradshaw-Pierce E, Cortes-Burgos L, Hall T, Johnston A, Murphy M, Nemirovskiy O, Ogawa S, Pegg L, Pelc M, Prinsen M, Schnute M, Wendling J, Wene S, Weinberg R, Wittwer A, Zweifel B, Masferrer J. J. Pharmacol. Exp. Ther. 2010; 334: 310
- 40 Miller LM, Keune W.-J, Castagna D, Young L, Duffy E, Potjewyd F, Salgado-Polo F, Engel García P, Semaan D, Pritchard J, Perrakis A, Macdonald SJ. F, Jamieson C, Watson AJ. B. J. Med. Chem. 2017; 60: 722
- 41a Hutchinson JH, Parr TA, Bunker KD, Lonergan D. WO 2015042053 A1, 2015
- 41b Hutchinson JH, Lonergan D. WO 2015077502 A1, 2015
- 41c Hutchinson JH, Parr TA, Bunker KD, Lonergan D. WO 2015042052 A1, 2015
- 41d Hutchinson JH, Lonergan D, Huang F, Rowbottom M, Calderon I. WO 2015048301 A1, 2015
- 41e Hutchinson JE, Lonegan D. WO 2015077503 A1, 2015
- 41f Stein AJ, Bain G, Prodanovich P, Santini AM, Darlington J, Stelzer NM. P, Sidhu RS, Schaub J, Goulet L, Lonergan D, Calderon I, Evans JF, Hutchinson JH. Mol. Pharmacol. 2015; 88: 982
- 42 Shah P, Cheasty A, Foxton C, Raynham T, Farooq M, Gutierrez IF, Lejeune A, Pritchard M, Turnbull A, Pang L, Owen P, Boyd S, Stowell A, Jordan A, Hamilton NM, Hitchin JR, Stockley M, MacDonald E, Quesada MJ, Trivier E, Skeete J, Ovaa H, Moolenaar WH, Ryder H. Bioorg. Med. Chem. Lett. 2016; 15: 5403
- 43a Castro CE, Gaughan EJ, Owsley DC. J. Org. Chem. 1966; 31: 4071
- 43b Sakamoto T, Kondo Y, Iwashita S, Nagano T, Yamanaka H. Chem. Pharm. Bull. 1988; 36: 1305
- 44a Humphrey GR, Kuethe JT. Chem. Rev. 2006; 106: 2875
- 44b Cacchi S, Fabrizi G. Chem. Rev. 2011; 111: PR215
- 45 Seath CP, Wilson KL, Campbell A, Mowat JM, Watson AJ. B. Chem. Commun. 2016; 52: 8703
- 46 Seath CP, Fyfe JW. B, Molloy JJ, Watson AJ. B. Synthesis 2017; 49: 891
- 47 Keune W.-J, Potjewyd F, Hediebrecht T, Salgado-Polo F, Macdonald SJ. F, Chelvarajan L, Latif AA, Soman S, Morris AJ, Watson AJ. B, Jamieson C, Perrakis A. J. Med. Chem. 2017; 60: 2006
- 48 Molloy JJ, Seath CP, West MJ, McLaughlin C, Fazakerley NJ, Kennedy AR, Nelson DJ, Watson AJ. B. J. Am. Chem. Soc. 2018; 140: 126
- 49 For some related work, see: Xu C, Fyfe JW. B, Seath CP, Bennett SH, Watson AJ. B. Chem. Commun. 2017; 53: 9139
- 50a Hunt AR, Stewart SK, Whiting A. Tetrahedron Lett. 1993; 34: 3599
- 50b Itami K, Tonogaki K, Nokami T, Ohashi Y, Yoshida J. Angew. Chem. Int. Ed. 2006; 45: 2404
- 50c Batsanov AS, Knowles JP, Whiting A. J. Org. Chem. 2007; 72: 2525
- 51a Amatore C, Jutand A, Le Duc G. Chem. Eur. J. 2011; 17: 2492
- 51b Amatore C, Jutand A, Le Duc G. Chem. Eur. J. 2012; 18: 6616
- 52a Thomas AA, Denmark SE. Science 2016; 352: 329
- 52b Thomas AA, Wang H, Zahrt AF, Denmark SE. J. Am. Chem. Soc. 2017; 139: 3805
- 53 For a review, see: Lennox AJ. J, Lloyd-Jones GC. Angew. Chem. Int. Ed. 2013; 52: 7362
- 54 Butters M, Harvey JN, Jover J, Lennox AJ. J, Lloyd-Jones GC, Murray PM. Angew. Chem. Int. Ed. 2010; 49: 5156
- 55 Cain DL, McLaughlin C, Molloy JJ, Carpenter-Warren C, Anderson NA, Watson AJ. B. Synlett 2019; 30: 787
For reviews, see:
For a selection of this work, see:
For reviews, see:
For examples, see:
For examples, see:
For examples, see:
For informative studies on the esterification of boronic acids, see:
For initial reports, see:
For selected reviews, see:
For selected examples, see:
-
References
- 1 Burns AR, Kerr JH, Kerr WJ, Passmore J, Paterson LC, Watson AJ. B. Org. Biomol. Chem. 2010; 8: 2777
- 2a Hayashi T, Yamasaki K. Chem. Rev. 2003; 103: 2829
- 2b Jean M, Casanova B, Gnoatto S, van de Weghe P. Org. Biomol. Chem. 2015; 13: 9168
- 3 For some of our collaborative medicinal chemistry work in Rh-catalyzed 1,4-addition, see: Anderson NA, Fallon BJ, Valverde E, MacDonald SJ. F, Pritchard JM, Suckling CJ, Watson AJ. B. Synlett 2012; 23: 2817
- 4 Rupnicki L, Saxena A, Lam HW. J. Am. Chem. Soc. 2009; 131: 10386
- 5 For a review, see: Best D, Lam HW. J. Org. Chem. 2014; 79: 831
- 6 Xu C, Muir CW, Leach AG, Kennedy AR, Watson AJ. B. Angew. Chem. Int. Ed. 2018; 57: 11374
- 7a MacMillan DS, Murray J, Sneddon HF, Jamieson C, Watson AJ. B. Green Chem. 2012; 14: 3016
- 7b MacMillan DS, Murray J, Sneddon HF, Jamieson C, Watson AJ. B. Green Chem. 2013; 15: 596
- 7c Caldwell N, Jamieson C, Simpson I, Watson AJ. B. ACS Sustain. Chem. Eng. 2013; 1: 1339
- 7d McGonagle FI, MacMillan DS, Murray J, Sneddon HF, Jamieson C, Watson AJ. B. Green Chem. 2013; 15: 1159
- 7e McGonagle FI, Sneddon HF, Jamieson C, Watson AJ. B. ACS Sustain. Chem. Eng. 2014; 2: 523
- 7f Caldwell N, Campbell PS, Jamieson C, Potjewyd F, Simpson I, Watson AJ. B. J. Org. Chem. 2014; 79: 9347
- 7g Caldwell N, Jamieson C, Simpson I, Watson AJ. B. Chem. Commun. 2015; 51: 9495
- 7h Eastman HE, Jamieson C, Watson AJ. B. Aldrichimica Acta 2015; 48: 51
- 7i Wilson KL, Kennedy AR, Murray J, Greatrex B, Jamieson C, Watson AJ. B. Beilstein J. Org. Chem. 2016; 12: 2005
- 7j McPherson CG, Caldwell N, Jamieson C, Simpson I, Watson AJ. B. Org. Biomol. Chem. 2017; 15: 3507
- 7k Wilson KL, Murray J, Sneddon HF, Wheelhouse KM. P, Watson AJ. B. Chem 2017; 3: 365
- 7l Wilson KL, Murray J, Jamieson C, Watson AJ. B. Synlett 2018; 29: 650
- 7m McPherson CG, Cooper AK, Bubliauskas A, Mulrainey P, Jamieson C, Watson AJ. B. Org. Lett. 2017; 19: 6736
- 7n Wilson L, Murray J, Jamieson C, Watson AJ. B. Org. Biomol. Chem. 2018; 16: 2851
- 7o Wilson KL, Murray J, Sneddon HF, Jamieson C, Watson AJ. B. Synlett 2018; 29: 2293
- 8a Barbayianni E, Magrioti V, Moutevelis-Minakakis GP, Kokotos G. Expert Opin. Ther. Pat. 2013; 23: 1123
- 8b Castagna D, Budd DC, Macdonald SJ. F, Jamieson C, Watson AJ. B. J. Med. Chem. 2016; 59: 5604
- 9a Swaney JS, Chapman C, Correa LD, Stebbins KJ, Bundey RA, Prodanovich PC, Fagan P, Baccei CS, Santini AM, Hutchinson JH, Seiders TJ, Parr TA, Prasit P, Evans JF, Lorrain DS. Br. J. Pharmacol. 2010; 160: 1699
- 9b Swaney JS, Chapman C, Correa LD, Stebbins KJ, Broadhead AR, Bain G, Santini AM, Darlington J, King CD, Baccei CS, Lee C, Parr TA, Roppe JR, Seiders TJ, Ziff J, Prasit P, Hutchinson JH, Evans JF, Lorrain DS. J. Pharmacol. Exp. Ther. 2011; 336: 693
- 10 Sinclair DJ, Sherburn MS. J. Org. Chem. 2005; 70: 3730
- 11 Dong C.-G, Hu Q.-S. J. Am. Chem. Soc. 2005; 127: 10006
- 12 Groombridge BJ, Goldup SM, Larrosa I. Chem. Commun. 2015; 51: 3832
- 13 Littke AF, Dai C, Fu GC. J. Am. Chem. Soc. 2000; 122: 4020
- 14 Almond-Thynne J, Blakemore DC, Prydeb DC, Spivey AC. Chem. Sci. 2017; 8: 40
- 15a Gillis EP, Burke MD. Aldrichimica Acta 2009; 42: 17
- 15b Li J, Grillo AS, Burke MD. Acc. Chem. Res. 2015; 48: 2297
- 16 Lee SJ, Gray KC, Paek JS, Burke MD. J. Am. Chem. Soc. 2008; 130: 466
- 17 Mancilla T, Contreras R, Wrackmeyer B. J. Organomet. Chem. 1986; 307: 1
- 18 Noguchi H, Hojo K, Suginome M. J. Am. Chem. Soc. 2007; 129: 758
- 19a Noguchi H, Shioda T, Chou C.-M, Suginome M. Org. Lett. 2008; 10: 377
- 19b Iwadate N, Suginome M. Org. Lett. 2009; 11: 1899
- 19c Iwadate N, Suginome M. Chem. Lett. 2010; 39: 558
- 19d Iwadate N, Suginome M. J. Am. Chem. Soc. 2010; 132: 2548
- 19e Ihara H, Koyanagi M, Suginome M. Org. Lett. 2011; 13: 2662
- 19f Feng X, Jeon H, Yun J. Angew. Chem. Int. Ed. 2013; 52: 3989
- 19g Yoshida H, Takemoto Y, Takaki K. Chem. Commun. 2015; 51: 6297
- 20 Endo K, Ohkubo T, Hirokami M, Shibata T. J. Am. Chem. Soc. 2010; 132: 11033
- 21a Mlynarski SN, Schuster CH, Morken JP. Nature 2014; 505: 386
- 21b Sun C, Potter B, Morken JP. J. Am. Chem. Soc. 2014; 136: 6534
- 21c Potter B, Szymaniak AA, Edelstein EK, Morken JP. J. Am. Chem. Soc. 2014; 136: 17918
- 22a Lee JC. H, McDonald R, Hall DG. Nature Chem. 2011; 3: 894
- 22b Sun H.-Y, Kubota K, Hall DG. Chem. Eur. J. 2015; 21: 19186
- 23a Crudden CM, Ziebenhaus C, Rygus JP. G, Ghozati K, Unsworth PJ, Nambo M, Both S, Hutchinson M, Laberge VS, Maekawa Y, Imao D. Nat. Commun. 2016; 7: 11065
- 23b Glasspoole BW, Oderinde MS, Moore BD, Antoft-Finch A, Crudden CM. Synthesis 2013; 45: 1759
- 24 Rygus JP. G, Crudden CM. J. Am. Chem. Soc. 2017; 139: 18124
- 26 Carrow BP, Hartwig JF. J. Am. Chem. Soc. 2011; 133: 2116
- 27 Fyfe JW. B, Valverde E, Seath CP, Kennedy AR, Redmond JM, Anderson NA, Watson AJ. B. Chem. Eur. J. 2015; 24: 8951
- 28 Knapp DM, Gillis EP, Burke MD. J. Am. Chem. Soc. 2009; 131: 6961
- 29a Babcock L, Pizer R. Inorg. Chem. 1980; 19: 56
- 29b Pizer R, Tihal CA. Polyhedron 1996; 15: 3411
- 29c Springsteen G, Wang B. Tetrahedron 2002; 58: 5291
- 29d Bosch LI, Fyles TM, James TD. Tetrahedron 2004; 60: 11175
- 29e Yan J, Springsteen G, Deeter S, Wang B. Tetrahedron 2004; 60: 11205
- 29f Iwatsuki S, Nakajima S, Inamo M, Takagi HD, Ishihara K. Inorg. Chem. 2007; 46: 354
- 29g Miyamoto C, Suzuki K, Iwatsuki S, Inamo M, Takagi HD, Ishihara K. Inorg. Chem. 2008; 47: 1417
- 29h Martínez-Aguirre MA, Villamil-Ramos R, Guerrero-Alvarez JA, Yatsmirsky AK. J. Org. Chem. 2013; 78: 4674
- 29i Watanabe E, Miyamoto C, Tanaka A, Iizuka K, Iwatsuki S, Inamo M, Takagi HD, Ishihara K. Dalton Trans. 2013; 42: 8446
- 29j Okamoto T, Tanaka A, Watanabe E, Miyazaki T, Sugaya T, Iwatsuki S, Inamo M, Takagi HD, Odani A, Ishihara K. Eur. J. Inorg. Chem. 2014; 2389
- 29k Furikado Y, Nagahata T, Okamoto T, Sugaya T, Iwatsuki S, Inamo M, Takagi HD, Odani A, Ishihara K. Chem. Eur. J. 2014; 20: 13194
- 30 Gonzalez JA, Maduka Ogba O, Morehouse GF, Rosson N, Houk KN, Leach AG, Cheong PH. Y, Burke MD, Lloyd-Jones GC. Nat. Chem. 2016; 8: 1067
- 31a Fyfe JW. B, Seath CP, Watson AJ. B. Angew. Chem. Int. Ed. 2014; 53: 12077
- 31b Molloy JJ, Law RP, Fyfe JW. B, Seath CP, Hirst DJ, Watson AJ. B. Org. Biomol. Chem. 2015; 13: 3093
- 32 The formal homologation also operates well for arylboronic acids, see: Muir CW, Vantourout JC, Isidro-Llobet A, Macdonald SJ. F, Watson AJ. B. Org. Lett. 2015; 17: 6030
- 33 Seath CP, Fyfe JW. B, Molloy JJ, Watson AJ. B. Angew. Chem. Int. Ed. 2015; 54: 9976
- 34 Fyfe JW. B, Fazakerley NJ, Watson AJ. B. Angew. Chem. Int. Ed. 2017; 56: 1249
- 35 Law RP, Atkinson SJ, Bamborough P, Chung C, Demont EH, Gordon LJ, Lindon M, Prinjha RK, Watson AJ. B, Hirst DJ. J. Med. Chem. 2018; 61: 4317
- 36 Castagna D, Duffy EL, Semann D, Young LC, Pritchard JM, MacDonald SJ. F, Budd DC, Jamieson C, Watson AJ. B. Med. Chem. Commun. 2015; 6: 1149
- 37a Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, Albers HM. H. G, van Meeteren LA, Houben AJ. S, van Zeijl L, Jansen S, Andries M, Hall T, Pegg LE, Benson TE, Kasiem M, Harlos K, Kooi CW. V, Smyth SS, Ovaa H, Bollen M, Morris AJ, Moolenaar WH, Perrakis A. Nat. Struct. Mol. Biol. 2011; 18: 198
- 37b Nishimasu H, Okudaira S, Hama K, Mihara E, Dohmae N, Inoue A, Ishitani R, Takagi J, Aoki J, Nureki O. Nat. Struct. Mol. Biol. 2011; 18: 205
- 37c Stein AJ, Bain G, Prodanovich P, Santini AM, Darlington J, Stelzer NM. P, Sidhu RS, Schaub J, Goulet L, Lonergan D, Calderon I, Evans JF, Hutchinson JH. Mol. Pharmacol. 2015; 88: 982
- 38a Roppe JR, Parr TA, Hutchinson JH. US 20130029948 A1, 2013
- 38b Hutchinson JH, Parr TA, Roppe JF, Stock NS, Volkots D. WO 2012024620 A2, 2012
- 38c Roppe JR, Parr TA, Hutchinson JH. WO 2012166415, 2012 .
- 39 Gierse J, Thorarensen A, Beltey K, Bradshaw-Pierce E, Cortes-Burgos L, Hall T, Johnston A, Murphy M, Nemirovskiy O, Ogawa S, Pegg L, Pelc M, Prinsen M, Schnute M, Wendling J, Wene S, Weinberg R, Wittwer A, Zweifel B, Masferrer J. J. Pharmacol. Exp. Ther. 2010; 334: 310
- 40 Miller LM, Keune W.-J, Castagna D, Young L, Duffy E, Potjewyd F, Salgado-Polo F, Engel García P, Semaan D, Pritchard J, Perrakis A, Macdonald SJ. F, Jamieson C, Watson AJ. B. J. Med. Chem. 2017; 60: 722
- 41a Hutchinson JH, Parr TA, Bunker KD, Lonergan D. WO 2015042053 A1, 2015
- 41b Hutchinson JH, Lonergan D. WO 2015077502 A1, 2015
- 41c Hutchinson JH, Parr TA, Bunker KD, Lonergan D. WO 2015042052 A1, 2015
- 41d Hutchinson JH, Lonergan D, Huang F, Rowbottom M, Calderon I. WO 2015048301 A1, 2015
- 41e Hutchinson JE, Lonegan D. WO 2015077503 A1, 2015
- 41f Stein AJ, Bain G, Prodanovich P, Santini AM, Darlington J, Stelzer NM. P, Sidhu RS, Schaub J, Goulet L, Lonergan D, Calderon I, Evans JF, Hutchinson JH. Mol. Pharmacol. 2015; 88: 982
- 42 Shah P, Cheasty A, Foxton C, Raynham T, Farooq M, Gutierrez IF, Lejeune A, Pritchard M, Turnbull A, Pang L, Owen P, Boyd S, Stowell A, Jordan A, Hamilton NM, Hitchin JR, Stockley M, MacDonald E, Quesada MJ, Trivier E, Skeete J, Ovaa H, Moolenaar WH, Ryder H. Bioorg. Med. Chem. Lett. 2016; 15: 5403
- 43a Castro CE, Gaughan EJ, Owsley DC. J. Org. Chem. 1966; 31: 4071
- 43b Sakamoto T, Kondo Y, Iwashita S, Nagano T, Yamanaka H. Chem. Pharm. Bull. 1988; 36: 1305
- 44a Humphrey GR, Kuethe JT. Chem. Rev. 2006; 106: 2875
- 44b Cacchi S, Fabrizi G. Chem. Rev. 2011; 111: PR215
- 45 Seath CP, Wilson KL, Campbell A, Mowat JM, Watson AJ. B. Chem. Commun. 2016; 52: 8703
- 46 Seath CP, Fyfe JW. B, Molloy JJ, Watson AJ. B. Synthesis 2017; 49: 891
- 47 Keune W.-J, Potjewyd F, Hediebrecht T, Salgado-Polo F, Macdonald SJ. F, Chelvarajan L, Latif AA, Soman S, Morris AJ, Watson AJ. B, Jamieson C, Perrakis A. J. Med. Chem. 2017; 60: 2006
- 48 Molloy JJ, Seath CP, West MJ, McLaughlin C, Fazakerley NJ, Kennedy AR, Nelson DJ, Watson AJ. B. J. Am. Chem. Soc. 2018; 140: 126
- 49 For some related work, see: Xu C, Fyfe JW. B, Seath CP, Bennett SH, Watson AJ. B. Chem. Commun. 2017; 53: 9139
- 50a Hunt AR, Stewart SK, Whiting A. Tetrahedron Lett. 1993; 34: 3599
- 50b Itami K, Tonogaki K, Nokami T, Ohashi Y, Yoshida J. Angew. Chem. Int. Ed. 2006; 45: 2404
- 50c Batsanov AS, Knowles JP, Whiting A. J. Org. Chem. 2007; 72: 2525
- 51a Amatore C, Jutand A, Le Duc G. Chem. Eur. J. 2011; 17: 2492
- 51b Amatore C, Jutand A, Le Duc G. Chem. Eur. J. 2012; 18: 6616
- 52a Thomas AA, Denmark SE. Science 2016; 352: 329
- 52b Thomas AA, Wang H, Zahrt AF, Denmark SE. J. Am. Chem. Soc. 2017; 139: 3805
- 53 For a review, see: Lennox AJ. J, Lloyd-Jones GC. Angew. Chem. Int. Ed. 2013; 52: 7362
- 54 Butters M, Harvey JN, Jover J, Lennox AJ. J, Lloyd-Jones GC, Murray PM. Angew. Chem. Int. Ed. 2010; 49: 5156
- 55 Cain DL, McLaughlin C, Molloy JJ, Carpenter-Warren C, Anderson NA, Watson AJ. B. Synlett 2019; 30: 787
For reviews, see:
For a selection of this work, see:
For reviews, see:
For examples, see:
For examples, see:
For examples, see:
For informative studies on the esterification of boronic acids, see:
For initial reports, see:
For selected reviews, see:
For selected examples, see: